Episode 8: 리소좀 축적질환 (LSD) — Variant보다 중요한 것은 패턴이다
Key Takeaways
- 리소좀 축적질환 (LSD)은 단일 장기 질환이 아니라 세포 내 분해 시스템 이상으로 발생하는 전신 질환군입니다.
- 리소좀 축적질환 (LSD) 판독에서는 질환명을 맞히는 것보다 표현형과 축적 물질의 패턴을 연결하는 것이 중요합니다.
- 표현형 → 침범 장기 → 축적 물질 → pathway → 후보 유전자 순서로 접근하면 interpretation이 더 구조화될 수 있습니다.
- Biomarker와 효소활성검사는 variant interpretation의 방향을 결정하는 중요한 근거가 됩니다.
- 리소좀 축적질환 (LSD)해석에서는 phenotype, biomarker, 효소 기능, disease mechanism을 하나의 causality chain으로 연결해야 합니다.
📌 Series Introduction
이 시리즈는 3billion의 임상 유전학 전문가가 직접 작성한 글입니다. AI 기반 유전변이 판독 도구가 보편화된 시대에, 자동화된 우선순위화 이후 판독자가 실제로 무엇을 이해하고 어떻게 판단해야 하는지에 초점을 맞춥니다.
각 편에서는 하나의 질환군을 중심으로, 유전적 기전과 임상 스펙트럼을 함께 살펴보며 변이를 “찾는 것”을 넘어 “설명하고 해석하는” 데 필요한 관점을 공유하고자 합니다.
왜 리소좀 축적질환(LSD) 판독에서는 “단서 연결”이 중요한가?
리소좀 축적질환(Lysosomal Storage Disorders, LSD)은 대표적인 lysosomal storage diseases 질환군으로, 다양한 lysosomal disorder phenotype을 보일 수 있는 희귀질환군입니다.
Gaucher disease, Fabry disease, Niemann-Pick disease, Tay-Sachs disease, MPS, Pompe disease처럼 잘 알려진 질환도 있지만, 실제 케이스는 항상 전형적인 모습으로 나타나지 않습니다.
간비장비대가 먼저 보일 수도 있고, 신경계 퇴행이 중심일 수도 있습니다. 어떤 경우에는 심장비대, 신장질환, 골격계 이상, 근력저하, 통증, 피부 병변처럼 서로 다른 장기 증상이 흩어져 나타납니다.
그래서 리소좀 축적질환 (LSD) 판독에서 중요한 질문은 단순히
“이 환자가 Fabry disease인가?”
“Gaucher disease인가?”
가 아닙니다.
먼저 물어야 할 질문은 이것입니다.
이 환자의 여러 증상들이 하나의 ‘축적 질환’으로 설명될 수 있는가?
리소좀 축적질환(LSD)은 왜 전신 질환으로 나타나는가?
리소좀은 세포 안에서 불필요하거나 손상된 물질을 분해하는 역할을 합니다. 하지만 특정 효소가 결핍되거나, 수송 단백질에 문제가 생기거나, 효소가 리소좀까지 제대로 이동하지 못하면 분해되어야 할 물질이 세포 안에 축적됩니다.
이 축적은 시간이 지나면서 세포 기능을 방해하고, 여러 장기 손상으로 이어집니다.
판독자 입장에서 중요한 점은 바로 여기에 있습니다.
리소좀 축적질환 (LSD)는 특정 장기 하나의 문제가 아니라, 세포 내 분해 시스템의 이상이 전신 표현형으로 나타나는 질환군입니다.
따라서 환자의 증상이 신경계, 심장, 간비장, 골격, 근육, 신장 등 여러 장기에 걸쳐 나타날수록, 단일 증상 중심의 해석보다는 “축적 질환 가능성”을 염두에 둔 접근이 필요합니다.
리소좀 축적질환 (LSD) 해석에서는 왜 “무엇이 축적되는가”가 중요한가?
리소좀 축적질환 (LSD)는 질환명도 많고 관련 유전자도 다양합니다. 하지만 판독 관점에서는 질환명을 하나씩 외우는 것보다 축적 물질 기준으로 구조화해서 보는 것이 더 유용합니다.
예를 들어 sphingolipid가 축적되면 Gaucher disease, Fabry disease, Tay-Sachs disease 같은 sphingolipidosis를 떠올릴 수 있습니다.
Glycosaminoglycan, 즉 GAG가 축적되면 MPS 계열 질환을 고려해야 합니다. Glycogen이 리소좀 안에 축적되면 Pompe disease가 주요 후보가 됩니다. Cholesterol 또는 lipid trafficking 이상이 의심되면 Niemann-Pick type C처럼 transporter defect를 생각해야 합니다.
즉, 판독 과정은 다음 흐름으로 정리할 수 있습니다.
리소좀 축적질환 (LSD) Interpretation Framework:
표현형 → 침범 장기 → 축적 물질 → 관련 pathway → 후보 유전자
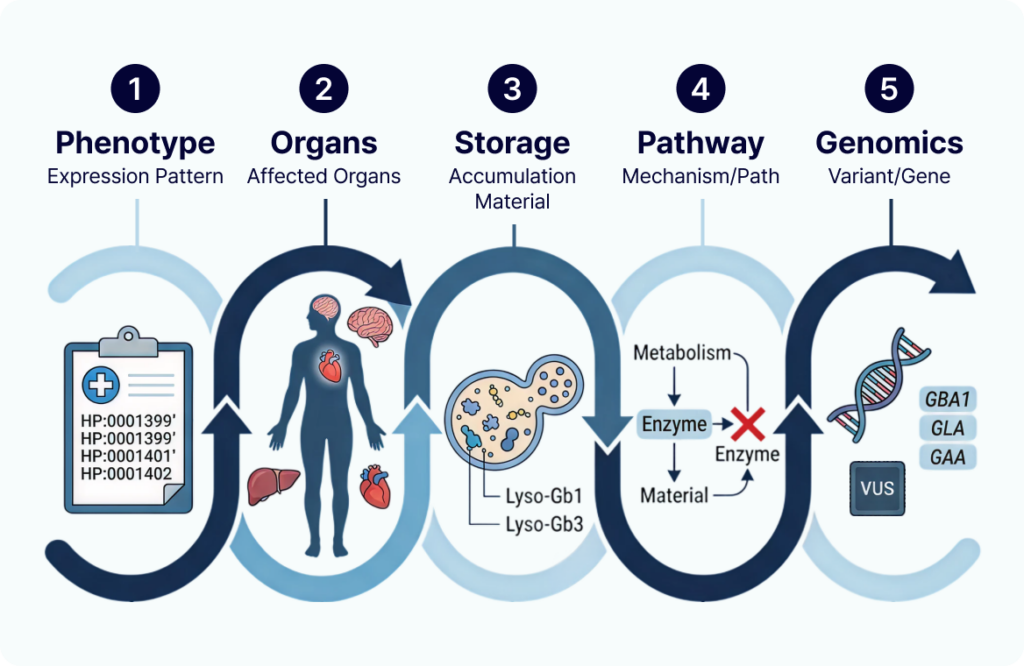
이 순서로 보면, 단순 variant filtering보다 훨씬 임상적으로 설명 가능한 후보를 좁혀갈 수 있습니다.
Gaucher, Fabry, Pompe 질환을 “패턴”으로 읽어야 하는 이유
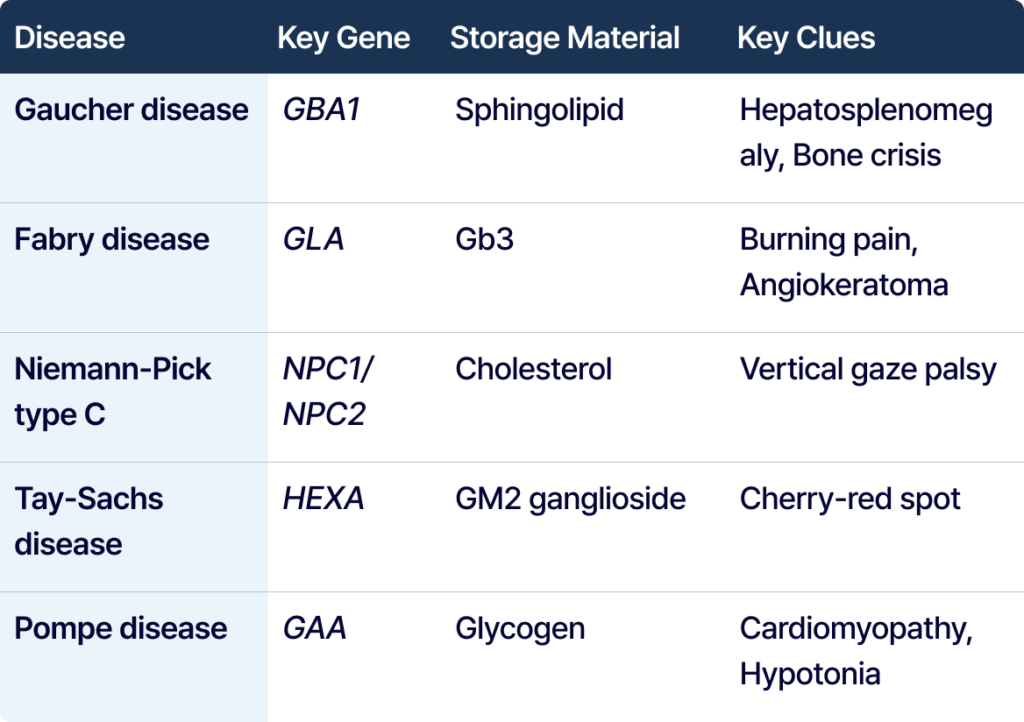
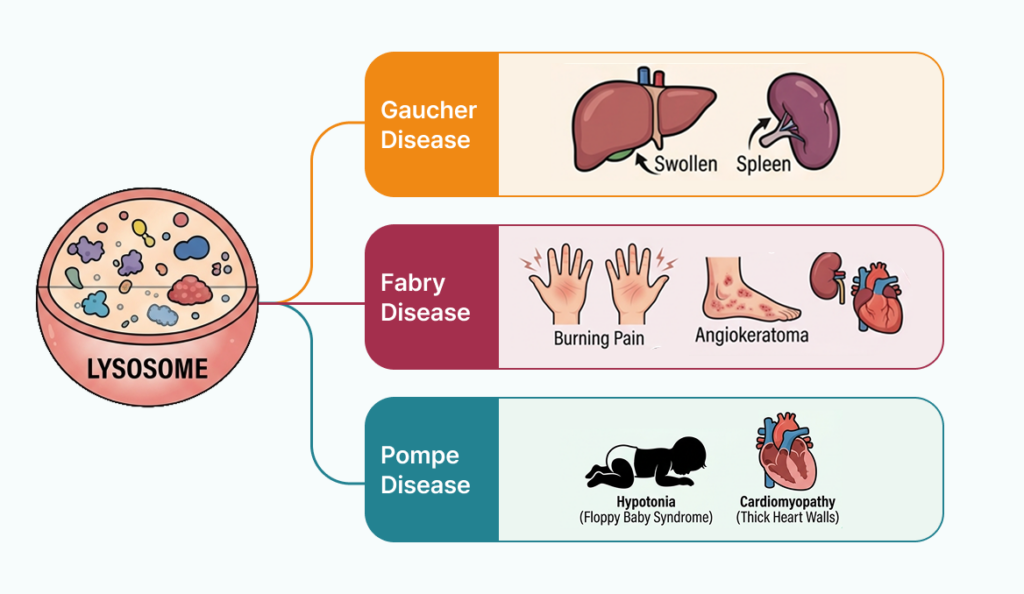
Gaucher disease는 GBA1 유전자와 관련되며, 간비장비대, 빈혈, 혈소판 감소, 골통증, bone crisis, 골괴사 등이 중요한 단서가 됩니다. 특히 간비장비대에 혈액학적 이상과 골 침범이 함께 보이면 GBA1을 주의 깊게 볼 필요가 있습니다.
Fabry disease는 GLA 유전자와 관련된 X-linked 질환입니다. 손발의 burning pain, angiokeratoma, hypohidrosis, 신장 침범, 심장 침범, 젊은 연령의 stroke risk가 주요 단서입니다. 다만 실제 판독에서는 성인 심근병증이나 신장질환으로 먼저 보일 수도 있어, Lyso-Gb3 같은 biomarker와 함께 해석하는 것이 중요합니다.
Niemann-Pick disease는 type A/B와 type C를 구분해야 합니다. Type A/B는 SMPD1과 관련된 acid sphingomyelinase 결핍이고, type C는 NPC1 또는 NPC2 이상으로 인한 cholesterol trafficking defect입니다. 특히 type C에서는 vertical gaze palsy, ataxia, cognitive decline, hepatosplenomegaly 같은 조합이 중요합니다.
Tay-Sachs disease는 HEXA 유전자와 관련되며, infantile neurodegeneration, 발달 퇴행, hyperacusis, cherry-red spot이 중요한 단서입니다.
MPS는 GAG 축적 질환군으로, coarse facial feature만 보는 것이 아니라 skeletal abnormality, short stature, joint contracture, organomegaly, airway issue, hearing loss, developmental delay 같은 전신 패턴을 함께 봐야 합니다.
Pompe disease는 GAA 유전자와 관련된 리소좀 glycogen 축적 질환입니다. Infantile-onset에서는 hypotonia, feeding difficulty와 hypertrophic cardiomyopathy가 중요하고, late-onset에서는 proximal muscle weakness, respiratory muscle weakness, exercise intolerance가 핵심 단서가 됩니다.
이처럼 리소좀 축적질환 (LSD) 판독에서는 개별 질환명을 아는 것보다, 환자의 표현형이 특정 축적 물질과 disease mechanism으로 설명되는지를 보는 것이 중요합니다.
왜 biomarker가 리소좀 축적질환 해석에서 중요한가?
리소좀 축적질환 (LSD)는 유전자검사만으로 결론 내리기 어려운 경우가 많습니다.
variant가 VUS로 나올 수도 있고, phenotype이 전형적이지 않을 수도 있습니다. 이때 biomarker는 해석의 방향을 잡는 중요한 근거가 됩니다.
대표적인 리소좀 축적질환 (LSD) biomarker 예시:
– Fabry disease → Lyso-Gb3
– Gaucher disease → Lyso-Gb1
– MPS → urinary GAG
– Pompe disease → enzyme activity assay
판독자는 유전자 변이, 표현형, biomarker, 효소활성검사 결과를 따로 보는 것이 아니라 하나의 causality chain으로 연결해야 합니다.
유전자 이상이 효소 기능 저하를 설명하는가?
효소 기능 저하가 특정 물질 축적을 설명하는가?
그 축적 물질이 환자의 장기 침범 패턴을 설명하는가?
이 세 가지 질문이 연결될 때, 리소좀 축적질환 (LSD) 판독의 설득력이 높아집니다.
왜 NGS 결과만으로 리소좀 축적질환 진단이 어려운가?
현재 리소좀 축적질환 (LSD) 진단에는 NGS panel, exome sequencing, genome sequencing이 폭넓게 활용됩니다. 특히 atypical phenotype, late-onset disease, overlapping presentation에서는 유전자검사가 진단 가능성을 크게 높여줍니다.
하지만 NGS 결과는 후보 변이를 보여줄 뿐, 그 자체로 결론을 만들어주지는 않습니다. 리소좀 축적질환 (LSD) 판독에서 중요한 것은 후보 변이를 찾는 것만이 아닙니다.
그 변이가 disease mechanism과 연결되는지, 환자의 phenotype을 설명하는지, biomarker 또는 효소활성검사 결과와 모순되지 않는지,
하나의 진단 가설로 충분히 방어 가능한지를 판단해야 합니다.
이 과정이 바로 희귀질환 판독자의 역할입니다.
GEBRA는 리소좀 축적질환 (LSD) interpretation workflow를 어떻게 지원하는가?
GEBRA는 rare disease interpretation을 위한 플랫폼으로, 판독자가 variant, gene, phenotype, disease evidence를 한 화면에서 연결해 볼 수 있도록 설계되었습니다. LSD처럼 phenotype이 여러 장기에 흩어져 있고, biomarker와 유전자 정보의 통합 해석이 중요한 질환군에서는 단순 variant filtering만으로는 충분하지 않습니다.
판독자는 다음 질문을 계속 확인해야 합니다.
- 이 variant가 disease mechanism과 맞는가?
- 환자의 HPO term과 질환 phenotype이 얼마나 잘 맞는가?
- 해당 gene-disease relationship은 충분히 설득력 있는가?
- VUS라도 보고 가능한 임상적 맥락이 있는가?
- 추가로 확인해야 할 biomarker 또는 functional evidence는 무엇인가?
GEBRA는 이러한 질문을 판독 과정 안에서 더 빠르고 일관되게 검토할 수 있도록 돕습니다.
마무리
리소좀 축적질환 판독은 질환명을 맞히는 과정이 아닙니다.
분해되지 못한 물질이 축적되고, 그 축적이 특정 장기 침범 패턴을 만들며, 그 패턴이 유전자와 biomarker를 통해 설명될 수 있는지를 확인하는 과정입니다. 그래서 리소좀 축적질환 (LSD) 케이스를 볼 때는 단일 증상보다 패턴을, 단일 변이보다 pathway를, 단일 검사 결과보다 전체 causality를 함께 봐야 합니다.
그 연결이 명확해질 때, 판독자는 단순한 후보 제시를 넘어 임상적으로 설명 가능한 진단 결론에 가까워질 수 있습니다.
직접 GEBRA에서 확인해보세요
미진단 케이스나 해석이 어려운 리소좀 축적질환 (LSD) 의심 케이스가 있다면, GEBRA에서 직접 판독해보실 수 있습니다.
Variant prioritization, phenotype matching, gene-disease evidence, ACMG evidence review를 한 흐름 안에서 확인하며, 기존 판독 과정과 비교해볼 수 있습니다.
GEBRA 데모를 신청하고, 실제 케이스에서 해석 과정이 어떻게 달라지는지 확인해보세요.
자주 묻는 질문 (FAQ)
Q1. 리소좀 축적질환(LSD)이란 무엇인가요?
리소좀 축적질환(Lysosomal Storage Disorders, LSD)은 리소좀 내 효소 또는 수송 시스템 이상으로 인해 분해되어야 할 물질이 세포 안에 축적되는 희귀질환군입니다. 이러한 축적은 시간이 지나면서 신경계, 심장, 간비장, 골격계, 근육, 신장 등 여러 장기에 영향을 미치며 다양한 전신 표현형으로 나타날 수 있습니다.
Q2. 왜 phenotype pattern이 리소좀 축적질환 판독에서 중요한가요?
리소좀 축적질환 (LSD)는 항상 전형적인 증상으로 나타나지 않기 때문입니다. 같은 질환이라도 환자마다 침범 장기와 증상 조합이 다를 수 있으며, 초기에는 특정 장기 증상만 보이는 경우도 많습니다. 따라서 판독자는 단일 증상보다 간비장비대, 신경계 이상, 심장 침범, 골격계 이상 같은 전체 phenotype pattern이 하나의 축적 질환으로 설명될 수 있는지를 함께 고려해야 합니다.
Q3. Biomarker는 리소좀 축적질환 해석에서 어떤 역할을 하나요?
Biomarker는 단순 보조 정보가 아니라 variant interpretation의 방향을 결정하는 중요한 근거가 됩니다. 예를 들어 Fabry disease에서는 Lyso-Gb3, Gaucher disease에서는 Lyso-Gb1, MPS에서는 urinary GAG가 중요한 단서가 될 수 있습니다. 판독자는 유전자 변이, biomarker, 효소활성검사, phenotype 정보를 하나의 causality chain으로 연결해 해석해야 합니다.
Q4. 왜 NGS 결과만으로 리소좀 축적질환 진단이 어려운가요?
NGS는 후보 변이를 찾는 데 매우 유용하지만, 그 자체만으로 질환을 확정할 수는 없습니다. 리소좀 축적질환 (LSD)에서는 variant가 실제 disease mechanism과 연결되는지, 환자의 phenotype과 일치하는지, biomarker 또는 효소활성검사 결과와 모순되지 않는지를 함께 확인해야 합니다. 따라서 희귀질환 판독에서는 variant filtering보다 phenotype, pathway, biomarker를 통합적으로 해석하는 과정이 더 중요합니다.
References
- Ferreira CR, Gahl WA. Lysosomal storage diseases. Transl Sci Rare Dis. 2017;2:1–71. doi:10.3233/TRD-160005.
- Hong X, Daiker J, Sadilek M, et al. Biomarker testing for lysosomal diseases: A technical standard of the ACMG. Genet Med. 2024;26(12):101245. PMID:39499245.
- Hall PL, Burlina A, Hahn S, et al. Measurement of lysosomal enzyme activities: A technical standard of the ACMG. Genet Med. 2022;24(7):1411–1418.
- Filocamo M, Morrone A. Biomarkers in Lysosomal Storage Diseases. Diseases. 2016;4(4):40. doi:10.3390/diseases4040040.
- Sanofi Genzyme. Gaucher Disease 101 — Lysosomal Storage Disorders.
- SSIEM ETAC 2024. Lecture 5 — Overview on Lysosomal Storage Disorders.
- Mapping Lysosomal Storage Disorders with Neurological Features by Cellular Pathways: Towards Precision Medicine. Curr Issues Mol Biol. 2025.
- Gelb MH, et al. Enzymatic screening and diagnosis of lysosomal storage diseases. CDC Stacks reference document.
- Burton BK, et al. Newborn screening for lysosomal storage diseases: current state, challenges, and implementation guidance.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the ACMG and AMP. Genet Med. 2015;17(5):405–424.
- Brain Sci. 2024;14(11):1085. doi:10.3390/brainsci14111085.
- Sanofi. Pompe Disease Patient Education Materials — Signs and Symptoms in IOPD/LOPD Patients.
- Sun A. Lysosomal storage disease overview. Ann Transl Med. 2018;6(24):476. doi:10.21037/atm.2018.11.39.
3billion 뉴스레터 구독자만을 위한
희귀질환 진단 최신 정보를 받아보세요.

3billion Inc.
희귀질환 환자들이 진단과 치료에서 외면받지 않는 세상을 만들기 위해 쓰리빌리언은 하나의 미션을 바라봅니다.