Episode 10: [가부키 증후군] 얼굴, 심장, 성장, 면역까지 모두 영향을 주는 ‘염색질병(Chromatinopathy)’의 모든 것
“우리 아이는 특징적인 얼굴 모양을 가지고 있으면서, 키가 잘 자라지 않고 발달도 조금 느린 편입니다. 왜 하나의 장기가 아니라 전신에 걸쳐 이런 다양한 증상이 나타나는 걸까요?”
많은 희귀 유전질환은 하나의 단백질 기능 이상으로 특정 장기에만 문제가 생기는 경우가 많습니다. 하지만 가부키 증후군(Kabuki Syndrome)은 다릅니다. 이 질환은 유전자 발현 자체를 총괄 조절하는 시스템인 후성유전학적 기전(Epigenetic machinery)이 망가지면서 발생하는 대표적인 염색질병(Chromatinopathy)이기 때문입니다.
오늘은 가부키 증후군의 원인 유전자인 KMT2D와 KDM6A가 몸 안에서 어떻게 작용하는지, 그리고 최신 임상 트렌드인 표현형 확장(Phenotypic Expansion) 정보까지 심층적으로 알아보겠습니다.
🔍 가부키 증후군의 대표 임상 증상 및 특징
KMT2D와 KDM6A는 모든 조직에서 발현되지만, 배아 발생과 성장 과정의 특정 시기 및 조직에서는 유전자 용량(dosage)에 매우 민감합니다. 이 시기에 haploinsufficiency가 발생하면 발달 프로그램이 교란되어 다양한 기관의 이상이 나타나며, 조직별 dosage sensitivity의 차이로 인해 환자마다 증상의 종류와 중증도가 다양하게 나타납니다.
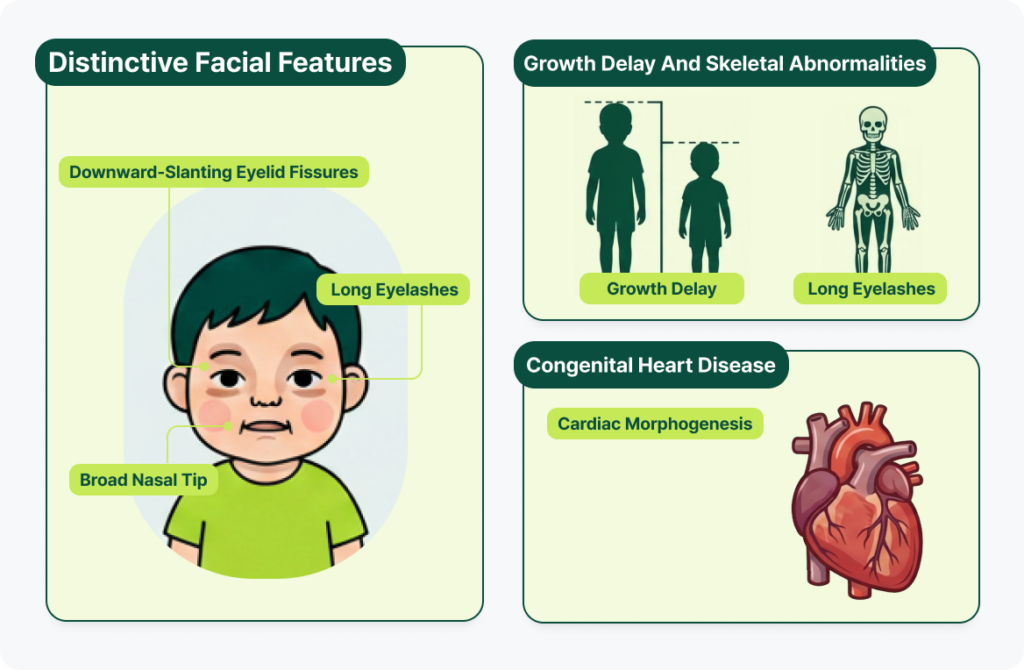
1. 신체적·외형적 특징
- 특징적인 안면 이상: 가부키 증후군이라는 이름의 유래가 될 정도로 눈꼬리가 길고 아래로 처진 눈, 긴 속눈썹, 넓은 콧등 등 특징적인 얼굴 형태를 보입니다. 이러한 얼굴 특징은 KMT2D/KDM6A 이상으로 인해 배아 발생 과정에서 신경능선 세포(neural crest cell)의 분화와 이동, 그리고 두개안면 발달을 조절하는 유전자 발현 프로그램이 교란되기 때문인 것으로 알려져 있습니다.
- 성장 지연 및 골격 기형: 출생 후 가파른 성장 정체를 겪으며 또래보다 키가 눈에 띄게 작아지는 성장 지연과 골격계 이상이 관찰됩니다.
- 선천성 심장질환: 신경능선 세포의 발달 장애 및 심장 형태 형성(Cardiac morphogenesis) 이상으로 인해 선천적인 심장 기형이 흔하게 동반됩니다.
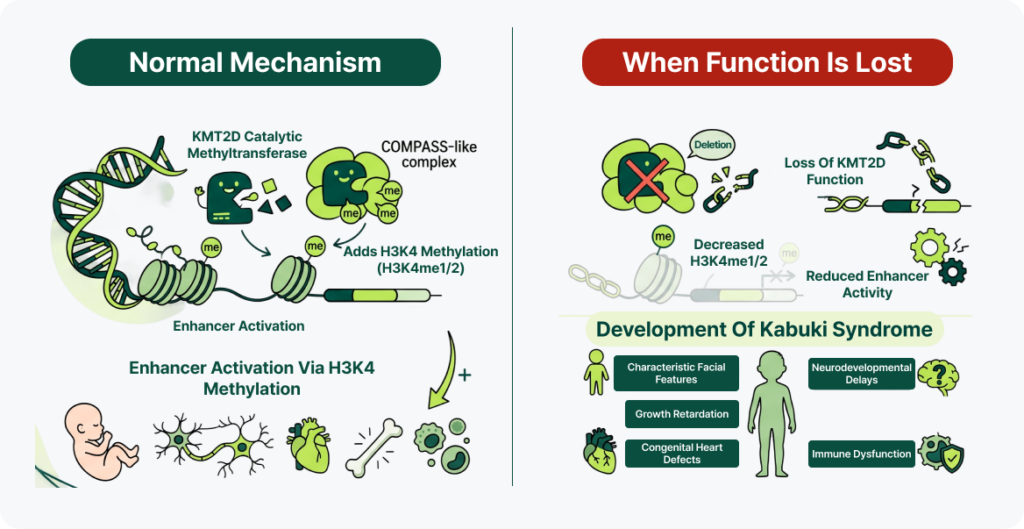
2. KMT2D 유전자의 역할: 켜기 표식 부여 (유전자 활성화)
- 정상 메커니즘: KMT2D는 COMPASS-like complex의 촉매(catalytic) methyltransferase로, H3K4에 methylation(H3K4me1/2)을 부가하여 enhancer를 활성화하고 발달에 필요한 유전자들의 발현을 조절합니다. KDM6A와 함께 chromatin remodeling과 조직 특이적인 유전자 발현을 조절하며, 배아 발생 과정에서 신경계, 심장, 골격 및 면역계의 정상적인 발달에 중요한 역할을 합니다.
- 기능 상실시: KMT2D 기능이 소실되면 enhancer 활성과 유전자 발현이 감소하여 발달 프로그램이 정상적으로 진행되지 못합니다. 이로 인해 골격 형성과 성장 조절, 해마 발달, 심장 형성(cardiogenesis), 그리고 B세포 및 T세포 분화가 교란되어 Kabuki syndrome의 특징적인 얼굴 형태, 성장지연, 신경 발달 이상, 선천성 심장기형 및 면역 이상이 발생합니다.
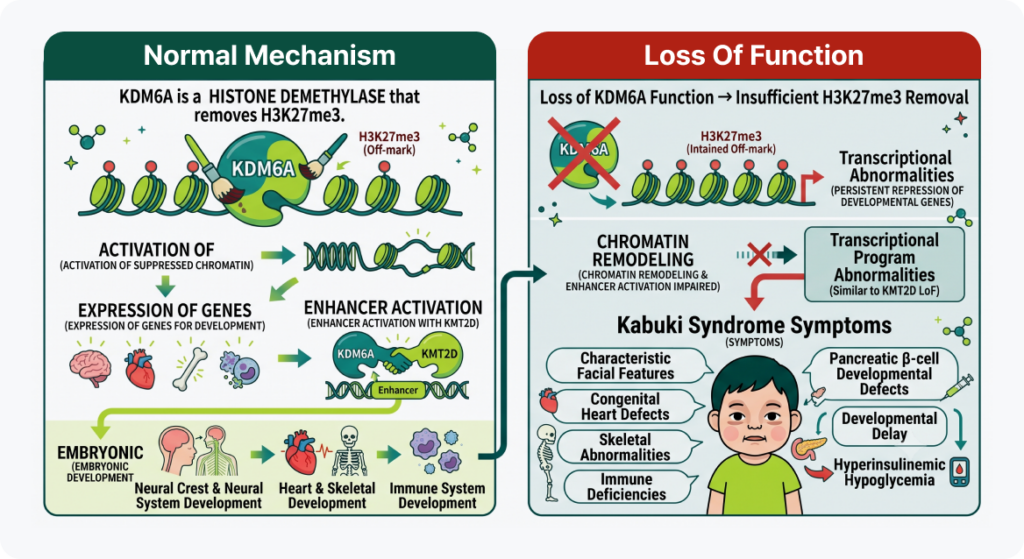
3. KDM6A 유전자의 역할: 끄기 표식 제거 (억제 해제)
- 정상 메커니즘: KDM6A는 H3K27me3를 제거하는 히스톤 탈메틸화 효소(histone demethylase)로, 억제된 chromatin을 활성화하여 발달에 필요한 유전자들이 정상적으로 발현되도록 합니다. 또한 KMT2D와 함께 enhancer를 활성화하며 조직 특이적인 유전자 발현을 조절하고, 배아 발생 과정에서 neural crest를 비롯한 신경계, 심장, 골격 및 면역계의 정상적인 발달에 중요한 역할을 합니다.
- 기능 상실(LoF) 시: KDM6A 기능이 소실되면 H3K27me3가 충분히 제거되지 않아 발달에 필요한 유전자들이 지속적으로 억제됩니다. 이로 인해 chromatin remodeling과 enhancer activation이 저해되어 KMT2D 기능 소실과 유사한 전사 프로그램 이상이 발생하며, neural crest를 비롯한 여러 발생 과정이 교란되어 특징적인 얼굴 형태, 선천성 심장기형, 발달지연, 골격 및 면역 이상 등 Kabuki syndrome의 다양한 임상 증상이 나타납니다. 또한 일부 환자에서는 췌장 β-cell 발달 이상과 관련된 hyperinsulinemic hypoglycemia가 동반될 수 있습니다.
변이 타입에 따른 임상적 변화: 표현형 확장 (Phenotypic Expansion)
기존 가부키 증후군은 유전자 기능을 완전히 잃어버리는 기능 소실(Loss-of-Function) 변이가 주를 이루었습니다. 그러나 최근 유전학 연구의 발전으로 매우 중요한 예외적 사실이 밝혀졌습니다.
KMT2D Exon 38/39 특정 미스센스 변이의 발견
최근 임상 데이터에 따르면, KMT2D 유전자의 exon 38 혹은 exon 39 영역에서 발생하는 특정 미스센스 변이(Missense variant)는 전형적인 가부키 증후군과는 완전히 다른 새로운 형태의 신체 증상을 유발한다는 것이 증명되었습니다. 이를 후성유전학에서는 표현형 확장(Phenotypic Expansion)의 대표 사례로 꼽습니다.
- 새롭게 관찰되는 주요 증상군: 후비공 폐쇄(Choanal atresia), 유두 형성 부전(Hypoplastic nipples), 부갑상선 기능 저하증(Hypoparathyroidism), 갑상선 기능 저하증(Hypothyroidism) 등 내분비 및 구조적 기형이 두드러집니다.
- 임상적 반전: 기존 Kabuki syndrome의 대표적인 특징인 전형적인 얼굴 형태와 지적장애는 현저히 경미하거나 비전형적으로 나타나는 경우가 많습니다.
- 시사점: 따라서 임상 현장에서는 단순히 특정 유전자의 변이 유무만 볼 것이 아니라, 변이가 일어난 정확한 위치(Exon 부위)와 변이 타입(LoF vs Missense)까지 함께 고려해야 정확한 확진과 조기 예방 치료가 가능합니다.
정밀 유전체 진단 플랫폼 GEBRA를 통한 정밀 분석
가부키 증후군은 단일 장기나 특정 구조의 이상으로 설명할 수 없는, 전신적인 유전자 발현 조절 실패(Gene expression dysregulation)로 인한 질환입니다. 이러한 복잡한 크로마틴 조절 기전을 규명하고 정밀의료의 새로운 길을 여는 중심에는 정밀 유전체 분석 솔루션 GEBRA가 있습니다.
GEBRA는 단순히 변이의 존재 여부를 확인하는 데 그치지 않고, KMT2D와 KDM6A 유전자의 특정 엑손 부위에서 어떤 변이 유형(LoF 또는 Missense)이 발생했는지를 고도화된 후성유전학적 데이터 레이어를 통해 정밀하게 분석합니다.
체계적인 맞춤 관리 가이드라인을 마련하고 싶으시다면, 지금 GEBRA의 고도화된 정밀 분석 서비스를 직접 경험해 보세요.
자주 묻는 질문 (FAQ)
Q1. 가부키 증후군은 자라면서 증상이 바뀔 수도 있나요?
A. 네, 그렇습니다. 가부키 증후군은 신체 골격 구조 자체가 고정되어 문제가 되는 구조 단백질 질환이 아니라, 유전자 발현을 실시간으로 켜고 끄는 조절 시스템의 질환(Epigenetic disorder)입니다. 따라서 아이의 연령대와 성장 단계에 따라 필요한 유전자 조절 양상이 달라지므로, 성장하면서 새로운 증상이 추가되거나 기존 표현형이 변할 수 있습니다. 지속적인 다학제적 모니터링이 필요한 이유가 바로 이 때문입니다.
Q2. KDM6A 변이는 X-linked(성염색체 유전)인데 왜 여성도 남성처럼 아픈가요?
A. KDM6A 유전자가 ‘X 염색체 불활성화’를 탈출(Escape)하기 때문입니다. 일반적인 여성은 두 개의 X 염색체 중 하나가 무작위로 잠기는(Inactivation) 과정을 거치므로 한쪽에 변이가 있어도 증상이 없거나 경미합니다. 하지만 KDM6A 유전자는 불활성화를 회피하여 여성에서도 양쪽 allele이 모두 정상 발현되어야만 하는 특성이 있습니다. 따라서 여성이라 하더라도 한쪽 유전자에만 기능 소실(Heterozygous LoF)이 발생하면 세포 내 단백질량이 부족해져 질환이 발병하게 됩니다. 다만, 대체적인 임상 표현형의 중증도는 남성 환자가 조금 더 심하게 나타나는 편입니다.
Q3. 가부키 증후군 원인 유전자인 KMT2D와 KDM6A의 구체적인 생물학적 원리는 무엇인가요?
A. 쉽게 비유하자면 KMT2D는 유전자를 켜기 위해 “활성화 표식(H3K4me)을 붙이는 효소”이고, KDM6A는 유전자가 닫히지 않도록 “억제 표식(H3K27me)을 지워버리는 지우개 효소”입니다. 두 효소의 분자생물학적 화학 작용은 정반대 방향 같지만, 결과적으로는 크로마틴(염색질) 구조를 느슨하게 열어주어 RNA 중합효소가 접근할 수 있는 환경을 유도한다는 점에서 완벽히 일치하는 발병 경로를 공유합니다.
Q4. 유전자 검사에서 가부키 증후군 변이가 나왔는데 얼굴 증상(안면 특징)이 전혀 없다면 오진일까요?
A. 오진이 아닐 가능성이 높습니다. 최근 규명된 표현형 확장(Phenotypic Expansion) 사례에 따르면, KMT2D 유전자의 특정 구간(Exon 38/39)에 발생하는 미스센스 변이 환자에서는 우리가 흔히 알고 있는 전형적인 Kabuki syndrome과 달리, 특징적인 얼굴 형태와 지적장애가 현저히 경미하거나 비전형적으로 나타나는 경우가 많습니다. 대신 갑상선 기능 저하증이나 후비공 폐쇄 같은 다른 장기의 기형이 주 증상으로 나타날 수 있으므로 , 변이의 상세 종류(Variant type)까지 정밀 분석하여 임상 증상과 매칭하는 것이 무엇보다 중요합니다.
References
Adam MP, Banka S, Bjornsson HT, et al. Kabuki syndrome: international consensus diagnostic criteria. J Med Genet. 2019;56(2):89–95.
Adam MP, Hudgins L, Hannibal M. Kabuki Syndrome. In: GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2013–2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK62111/
Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T. Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr. 1981;99(4):565–569.
Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr. 1981;99(4):570–573.
Cuvertino S, et al. A restricted spectrum of missense KMT2D variants cause a phenotype distinct from Kabuki syndrome. Genet Med. 2020;22(5):867–877.
Lee SY, Kim JH, Han SH, et al. Kabuki syndrome: clinical and molecular characteristics. Korean J Pediatr. 2015;58(8):317–324.
3billion 뉴스레터 구독자만을 위한
희귀질환 진단 최신 정보를 받아보세요.

3billion Inc.
희귀질환 환자들이 진단과 치료에서 외면받지 않는 세상을 만들기 위해 쓰리빌리언은 하나의 미션을 바라봅니다.