Episode 10: [Kabuki Syndrome] Everything About the “Chromatinopathy” That Affects the Face, Heart, Growth, and Immune System
“My child has distinctive facial features, isn’t growing as tall as expected, and seems to be developing a bit more slowly than other children. Why would so many different symptoms appear across the whole body instead of just one organ?”
Many rare genetic disorders arise from a defect in a single protein that affects only one specific organ. Kabuki syndrome is different. This condition is a representative chromatinopathy that occurs when the epigenetic machinery — the system that governs gene expression itself — breaks down.
Today, we take an in-depth look at how the causative genes of Kabuki syndrome, KMT2D and KDM6A, function in the body, along with the latest clinical trend of phenotypic expansion.
🔍 Representative Clinical Symptoms and Features of Kabuki Syndrome
KMT2D and KDM6A are expressed in nearly all tissues, but certain tissues are especially sensitive to gene dosage during specific windows of embryonic development and growth. When haploinsufficiency occurs during these windows, developmental programs are disrupted, and because dosage sensitivity varies by tissue, the type and severity of symptoms vary from patient to patient.
1. Physical and Facial Features
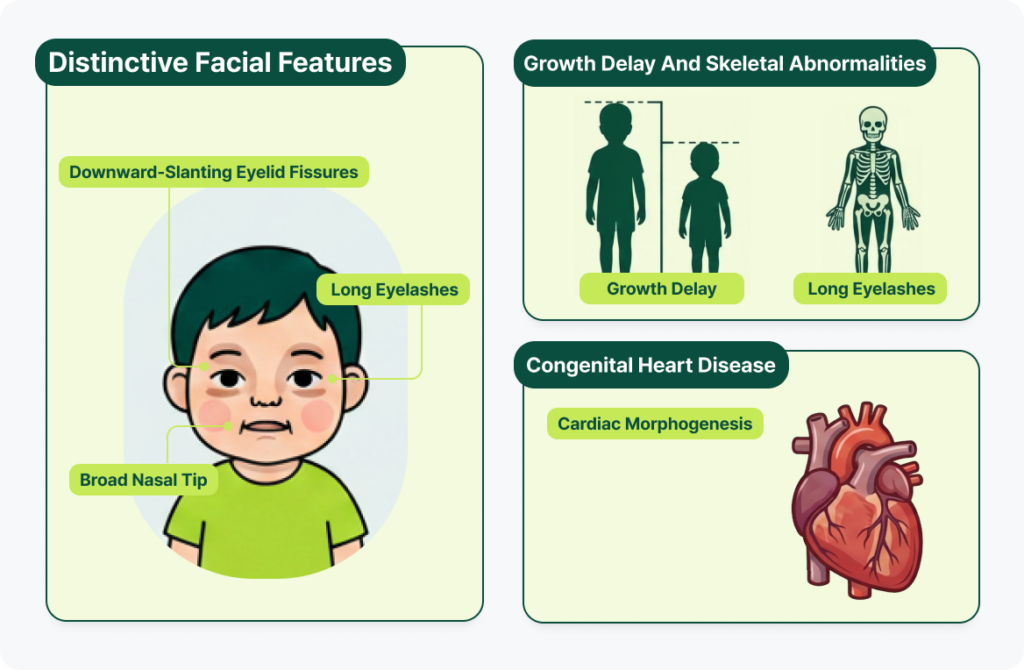
- Distinctive facial features: The features that gave the syndrome its name include long, downward-slanting eyelid fissures, long eyelashes, and a broad nasal tip. These facial characteristics are thought to result from disrupted gene-expression programs that normally govern the differentiation and migration of neural crest cells and craniofacial development — disruptions caused by KMT2D/KDM6A abnormalities.
- Growth delay and skeletal abnormalities: Patients often experience a sharp growth deceleration after birth, resulting in noticeably short stature compared to peers, along with skeletal abnormalities.
- Congenital heart disease: Congenital heart defects are common, arising from impaired neural crest cell development and abnormalities in cardiac morphogenesis.
2. Role of the KMT2D Gene: Adding the “On” Mark (Gene Activation)
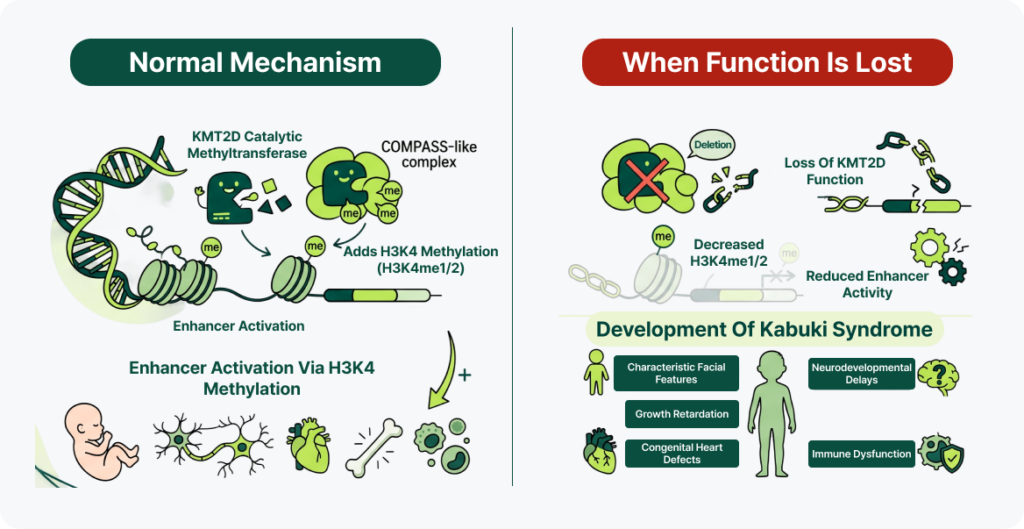
- Normal mechanism: KMT2D is the catalytic methyltransferase of the COMPASS-like complex. It adds methylation (H3K4me1/2) to H3K4, activating enhancers and regulating the expression of genes required for development. Together with KDM6A, it regulates chromatin remodeling and tissue-specific gene expression, playing a key role in the normal development of the nervous system, heart, skeleton, and immune system during embryogenesis.
- When function is lost: Loss of KMT2D function reduces enhancer activity and gene expression, preventing developmental programs from proceeding normally. This disrupts skeletal formation and growth regulation, hippocampal development, cardiogenesis, and B-cell/T-cell differentiation, producing the characteristic facial features, growth delay, neurodevelopmental abnormalities, congenital heart defects, and immune abnormalities seen in Kabuki syndrome.
3. Role of the KDM6A Gene: Removing the “Off” Mark (Derepression)
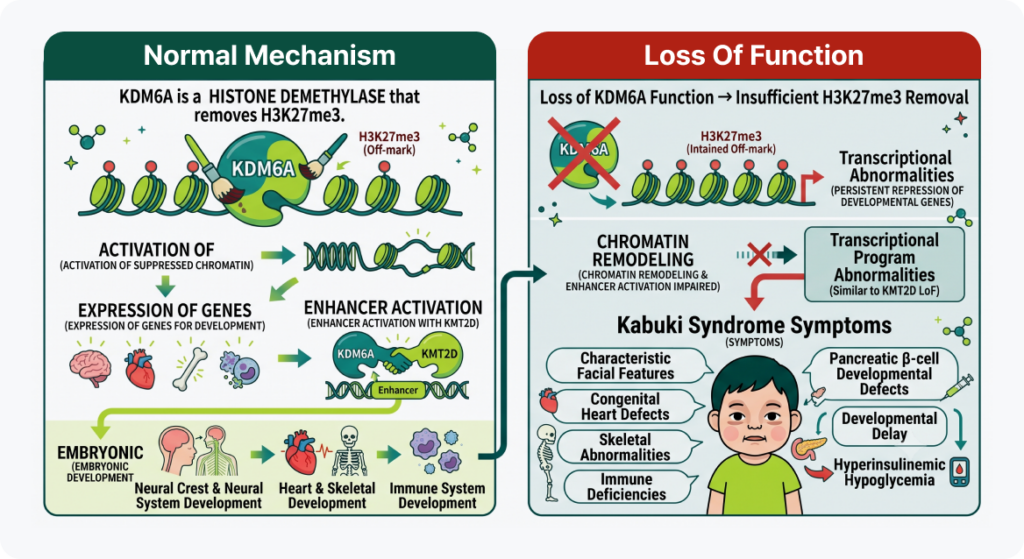
- Normal mechanism: KDM6A is a histone demethylase that removes H3K27me3, reactivating repressed chromatin so that genes required for development can be expressed normally. Along with KMT2D, it activates enhancers and regulates tissue-specific gene expression, playing an important role in the normal development of the neural crest and the nervous system, heart, skeleton, and immune system during embryogenesis.
- When loss-of-function (LoF) occurs: When KDM6A function is lost, H3K27me3 is not adequately removed, so genes required for development remain repressed. This impairs chromatin remodeling and enhancer activation, producing transcriptional abnormalities similar to those seen with KMT2D loss. Disruption of neural crest and other developmental processes leads to the characteristic facial features, congenital heart defects, developmental delay, and skeletal/immune abnormalities of Kabuki syndrome. Some patients also experience hyperinsulinemic hypoglycemia related to abnormal pancreatic β-cell development.
Clinical Variation by Variant Type: Phenotypic Expansion
Classic Kabuki syndrome has historically been driven mainly by loss-of-function variants that completely abolish gene function. However, recent advances in genetics have revealed an important exception.
Discovery of Specific Missense Variants in KMT2D Exon 38/39
Recent clinical data show that specific missense variants occurring in exon 38 or exon 39 of the KMT2D gene produce a distinct set of physical symptoms entirely different from classic Kabuki syndrome. In epigenetics, this is considered a leading example of phenotypic expansion.
- Newly observed symptom cluster: Endocrine and structural abnormalities stand out, including choanal atresia, hypoplastic nipples, hypoparathyroidism, and hypothyroidism.
- A clinical reversal: The classic facial features and intellectual disability typically associated with Kabuki syndrome are often mild or atypical in these patients.
- Clinical implication: In clinical practice, it is not enough to simply confirm the presence of a variant in a given gene — the exact location (exon) and the type of variant (LoF vs. missense) must also be considered to reach an accurate diagnosis and enable early preventive care.
Precision Genomic Analysis with the GEBRA Variant Interpretation Platform
Kabuki syndrome cannot be explained by an abnormality in a single organ or structure — it is a disorder rooted in systemic dysregulation of gene expression. At the center of unraveling this complex chromatin-regulation mechanism and advancing precision medicine is GEBRA, a precision genomic analysis solution.
GEBRA goes beyond simply confirming whether a variant is present. Using an advanced epigenetic data layer, it precisely analyzes which type of variant (LoF or missense) has occurred, and in which exon, within the KMT2D and KDM6A genes.
If you’d like to build a systematic, personalized management plan, experience GEBRA’s advanced precision analysis service for yourself today.
FAQ
Q1. Can the symptoms of Kabuki syndrome change as a child grows?
A. Yes. Kabuki syndrome is not a structural protein disorder in which the skeletal structure itself is fixed and problematic — it is an epigenetic disorder affecting the system that turns gene expression on and off in real time. Because the gene-regulation needs of the body change with age and developmental stage, new symptoms can emerge or existing features can change as the child grows. This is exactly why ongoing multidisciplinary monitoring is necessary.
Q2. KDM6A variants are X-linked — so why do females get sick just like males?
A. Because the KDM6A gene escapes X-chromosome inactivation. In most women, one of the two X chromosomes is randomly silenced, so a variant on one copy typically causes no or mild symptoms. However, the KDM6A gene evades this inactivation, meaning both alleles normally need to be expressed even in females. As a result, a loss-of-function variant in just one copy (heterozygous LoF) can reduce cellular protein levels enough to cause disease. That said, on average, male patients tend to show somewhat more severe clinical phenotypes.
Q3. Exactly how do the KMT2D and KDM6A genes open up chromatin?
A. Think of KMT2D as an enzyme that “attaches an activation mark (H3K4me)” to switch a gene on, and KDM6A as an “eraser enzyme” that removes a repression mark (H3K27me) to keep a gene from closing. Though their chemical actions point in opposite directions, both ultimately loosen the chromatin structure to create an environment RNA polymerase can access — meaning they converge on the same disease pathway.
Q4. My genetic test shows a Kabuki syndrome variant, but my child has none of the facial features. Could this be a misdiagnosis?
A. It’s unlikely to be a misdiagnosis. According to recently identified cases of phenotypic expansion, patients with specific missense variants in KMT2D exon 38/39 often show mild or atypical versions of the classic facial features and intellectual disability usually associated with Kabuki syndrome. Instead, abnormalities in other organs — such as hypothyroidism or choanal atresia — may be the primary presentation. This makes it especially important to analyze the precise variant type in detail and match it against the clinical picture.
References
Adam MP, Banka S, Bjornsson HT, et al. Kabuki syndrome: international consensus diagnostic criteria. J Med Genet. 2019;56(2):89–95.
Adam MP, Hudgins L, Hannibal M. Kabuki Syndrome. In: GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2013–2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK62111/
Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T. Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr. 1981;99(4):565–569.
Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr. 1981;99(4):570–573.
Cuvertino S, et al. A restricted spectrum of missense KMT2D variants cause a phenotype distinct from Kabuki syndrome. Genet Med. 2020;22(5):867–877.
Lee SY, Kim JH, Han SH, et al. Kabuki syndrome: clinical and molecular characteristics. Korean J Pediatr. 2015;58(8):317–324.
Get exclusive rare disease updates
from 3billion.

3billion Inc.
3billion is dedicated to creating a world where patients with rare diseases are not neglected in diagnosis and treatment.