Five Clinical Red Flags for Suspecting hATTR in Patients

When evaluating patients who present with both peripheral neuropathy and cardiomyopathy, the possibility of hereditary transthyretin (hATTR) amyloidosis must always be considered in the differential diagnosis.
hATTR amyloidosis is a condition in which an average of 5.8 years elapses from symptom onset to confirmed diagnosis (Bhadola et al., 2020). Throughout this period, TTR amyloid accumulates irreversible damage in the nervous system and the heart. Because involvement of each organ can readily be attributed to more common, standalone diagnoses, failure to recognize the multisystem pattern as a unified clinical entity perpetuates diagnostic delay.
Early recognition determines when treatment can be initiated, and the timing of treatment initiation determines prognosis. This post outlines five actionable red flags for hATTR amyloidosis and a diagnostic approach for definitive confirmation.
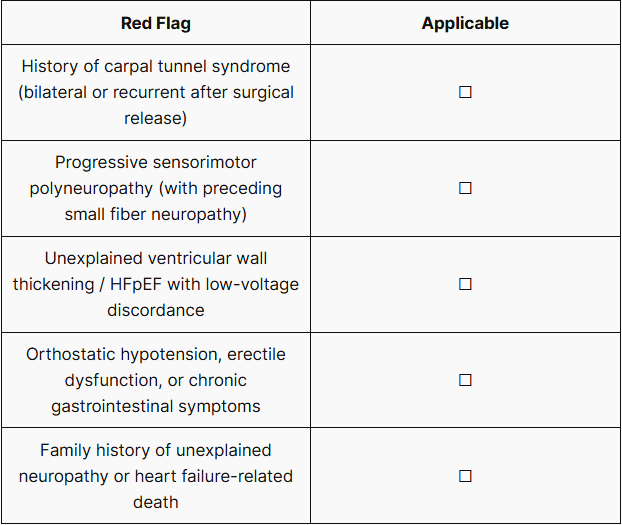
🔴 Red Flag 1. Carpal Tunnel Syndrome — An Early Signal Preceding Diagnosis by Up to 9 Years
In hATTR amyloidosis, carpal tunnel syndrome (CTS) is a characteristic early finding that appears significantly earlier than overt neuropathy or cardiomyopathy. Evidence indicates that CTS precedes the final diagnosis of hATTR by an average of 5 to 9 years, with a notably higher prevalence among patients with cardiac amyloidosis compared to the general population (Milandri et al., 2020; Bhadola et al., 2020).
One particularly noteworthy dataset warrants attention. Histopathological analysis of tenosynovial tissue obtained during CTS release surgery reveals Congo Red positivity in approximately 10% of specimens (positivity rates may vary by population), and of these, approximately 20% are ultimately diagnosed with hATTR (Bhadola et al., 2020). This means that the operative setting of CTS surgery represents a critical — and largely underutilized — opportunity for early detection of hATTR amyloidosis. While CTS alone is not sufficient for definitive diagnosis, it is unequivocally an important clinical signal.
Clinical Pearls
- Bilateral CTS, or CTS with symptom recurrence following surgical release, should always prompt suspicion for hATTR
- Musculoskeletal manifestations such as lumbar spinal stenosis and spontaneous tendon rupture may also precede the diagnosis
- Histological examination of tenosynovial tissue with Congo Red staining and neurophysiological studies should be actively considered at the time of CTS surgery (Bhadola et al., 2020)
🔴 Red Flag 2. Progressive Sensorimotor Polyneuropathy
hATTR neuropathy is characterized by a complex pattern of sensory, motor, and autonomic involvement. Notably, small fiber neuropathy manifests early in the disease course, presenting with pain and sensory disturbance, and subsequently progresses to motor weakness.
The key distinguishing feature from diabetic or alcoholic neuropathy is the concurrent involvement of the autonomic nervous system and the heart. In hATTR, neuropathy rarely occurs in isolation; it tends to progress alongside autonomic dysfunction and cardiomyopathy. A positive Romberg sign serves as a supporting clinical finding that further raises suspicion for hATTR (Carroll et al., 2022).
Diagnostic Approach
- Nerve conduction studies (NCS) to characterize the sensorimotor neuropathy pattern
- Skin biopsy to assess intraepidermal nerve fiber density for small fiber involvement
- When small fiber neuropathy is confirmed, cardiac evaluation and genetic testing should be pursued in parallel
🔴 Red Flag 3. Unexplained Cardiomyopathy — Ventricular Wall Thickening in the Absence of Hypertension
Unexplained hypertrophic cardiomyopathy should prompt active consideration of TTR amyloid deposition. The characteristic cardiac findings of hATTR cardiomyopathy include the following:
- Ventricular wall thickening — particularly increased interventricular septum (IVS) thickness — occurring in the absence of hypertension
- A discordance between a heart failure with preserved ejection fraction (HFpEF) clinical picture and low-voltage findings on electrocardiography (i.e., hypertrophy on imaging with low voltage on ECG)
- Concurrent atrial fibrillation
- Disproportionate elevation of NT-proBNP/BNP relative to the apparent clinical severity
When this constellation of findings is observed, evaluation for cardiac amyloidosis should be pursued. It is important to note that echocardiographic findings alone may be insufficient to differentiate hATTR cardiomyopathy from AL amyloidosis; bone scintigraphy (99mTc-PYP/DPD) in conjunction with genetic testing is therefore essential.
🔴 Red Flag 4. Autonomic Dysfunction
Autonomic nervous system involvement in hATTR amyloidosis appears early in the disease course and manifests across multiple organ systems. Key symptoms include the following:
- Orthostatic hypotension: blood pressure drop upon standing, accompanied by dizziness
- Erectile dysfunction: frequently attributed to urological or endocrine causes, leading to missed diagnosis
- Gastrointestinal symptoms: unexplained chronic diarrhea, gastroparesis, and unintentional weight loss
When each symptom presents in isolation, it is commonly explained by a different etiology across different subspecialties. The clinical imperative is to recognize these symptoms collectively as a pattern of multisystem autonomic dysfunction. In patients with concurrent neuropathy or cardiomyopathy, the presence of autonomic symptoms must prompt consideration of hATTR amyloidosis.

🔴 Red Flag 5. Family History — But Absence of Family History Does Not Exclude the Diagnosis
hATTR amyloidosis follows an autosomal dominant inheritance pattern. A family history of unexplained neuropathy or heart failure-related death in a first-degree relative constitutes strong grounds for clinical suspicion. The distribution of pathogenic TTR variants differs by ethnic background:
- p.Val30Met (Val30Met): high frequency in Asian (Japanese) and Portuguese populations
- p.Val142Ile (Val142Ile / V122I): high frequency in individuals of African descent; predominantly manifests as the cardiomyopathy phenotype
However, de novo variants have been documented, and therefore the absence of a family history does not exclude hATTR amyloidosis (Carroll et al., 2022). Regardless of family history, genetic testing should be actively considered whenever the multisystem clinical pattern is consistent with hATTR.
✅ hATTR Red Flag Self-Assessment Checklist
If one or more of the following items apply, genetic testing for hATTR amyloidosis should be considered.
Genetic testing is a critical step in the definitive diagnosis of hATTR amyloidosis. Given the broad phenotypic variability of TTR variants and the existence of numerous rare pathogenic mutations, whole genome sequencing (WGS) or whole exome sequencing (WES) offers the following clinical advantages over single-gene testing (Carroll et al., 2022; Adams et al., 2020):
- Comprehensive coverage of the full spectrum of TTR variants, including rare variants and variants of uncertain significance (VUS)
- Simultaneous screening for other hereditary conditions presenting with similar phenotypes, when the differential diagnosis remains unclear
- Provision of a genomic dataset that simultaneously supports cascade screening of at-risk family members at the time of diagnosis
If genetic testing is required for the confirmation of suspected hATTR amyloidosis in your patient, please submit your inquiry via the button below. A member of our team will contact you promptly.
References
- Bhadola S, Lau K, Kaku M. Hereditary Transthyretin Amyloidosis and Carpal Tunnel Syndrome: Update on Opportunities for Earlier Diagnosis of Amyloidosis. Neurology. 2020.
- Milandri A, et al. Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies.2020.
- Carroll A, et al. Novel approaches to diagnosis and management of hereditary transthyretin amyloidosis. J Neurol Neurosurg Psychiatry. 2021.
- Adams D, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2021.
- Zhang et al. (2025), Peripheral Neuropathy in p.Val142Ile (Val122Ile) Variant Hereditary Transthyretin-Mediated Amyloidosis.2025.
Get exclusive rare disease updates
from 3billion.

3billion Inc.
3billion is dedicated to creating a world where patients with rare diseases are not neglected in diagnosis and treatment.