Episode 3 Ehlers-Danlos Syndrome (EDS): Beyond the Variant
Episode 3 Ehlers-Danlos Syndrome (EDS): Causes, Genetics, Molecular Mechanisms, and Clinical Features
📌 Series Introduction
In an era where AI-based variant interpretation tools have become widely adopted, this series focuses on what human interpreters must still understand and decide after automated prioritization.
Each episode centers on a specific disease group, examining genetic mechanisms and clinical spectra together, with the goal of moving beyond simply “finding variants” toward explaining and interpreting them in a clinically meaningful way.
This article provides a comprehensive overview of EDS definition, classification, genetic causes, molecular pathophysiology, and clinical diagnosis.
In Episode 3, we examine Ehlers-Danlos Syndrome (EDS)
Episode 3 — Ehlers-Danlos Syndrome (EDS)
🔍 What Is Ehlers-Danlos Syndrome?
Ehlers-Danlos syndrome is a group of rare inherited connective tissue disorders characterized by structural defects in connective tissues.
The syndrome is named after two dermatologists:
- Edvard Ehlers
- Henri-Alexandre Danlos
The core clinical features of EDS include:
Key Clinical Features
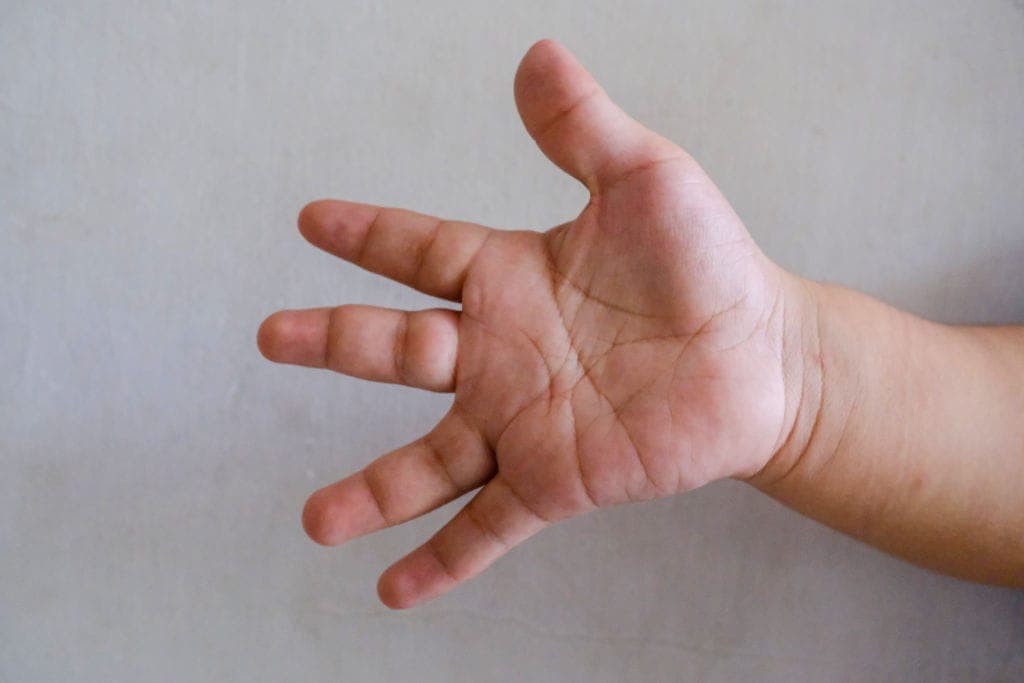
- Joint hypermobility
- Skin hyperextensibility
- Abnormal wound healing
- Easy bruising
Because connective tissue is widely distributed throughout the body, EDS is considered a multisystem disorder, affecting multiple organs and tissues rather than a single system.
🧬 Classification of Ehlers-Danlos Syndrome
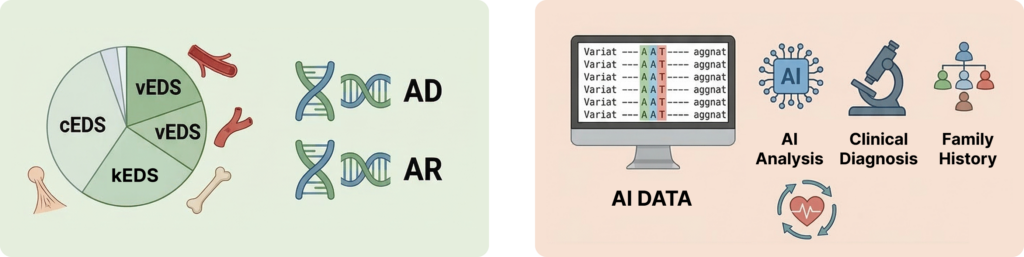
Currently, 14 clinical subtypes of EDS are recognized.
Some of the most well-known subtypes include:
- Classical EDS (cEDS)
- Vascular EDS (vEDS)
- Kyphoscoliotic EDS (kEDS)
- Myopathic EDS (mEDS)
- Periodontal EDS (pEDS)
While the genetic causes for 13 subtypes have identified molecular causes, Hypermobile EDS (hEDS) is the only type whose molecular cause remains unclear.
Prevalence of EDS
The prevalence varies by subtype:
- Classical EDS: approximately 1 in 20,000
- Vascular EDS: approximately 1 in 50,000–200,000
Several subtypes therefore fall within the category of rare diseases.
Genetic Causes of Ehlers-Danlos Syndrome
To date, around 20 genes have been associated with EDS.
Major genes include:
| COL5A1 | COL5A2 | COL1A1 | COL3A1 | |||
| COL1A2 | ADAMTS2 | PLOD1 | FKBP14 | |||
| TNXB | COL12A1 | CHST14 | DSE | |||
| B4GALT7 | B3GALT6 | SLC39A13 | ZNF469 | |||
| PRDM5 | C1R | C1S | AEBP1 |
EDS can be inherited through:
- Autosomal dominant inheritance
- Autosomal recessive inheritance
These EDS-related genes are broadly classified into three categories based on the function of the proteins they encode.
1. Fibrillar collagen proteins
- COL1A1
- COL3A1
- COL5A1
- COL5A2
2. Collagen processing enzymes
- ADAMTS2
- PLOD1
- FKBP14
3. ECM and proteoglycan-related proteins
- TNXB
- B4GALT7
- CHST14
- DSE
Variants in these genes ultimately disrupt collagen structure or extracellular matrix integrity.
Collagen Structure and Molecular Mechanisms in EDS
Understanding EDS requires an understanding of basic collagen biology.
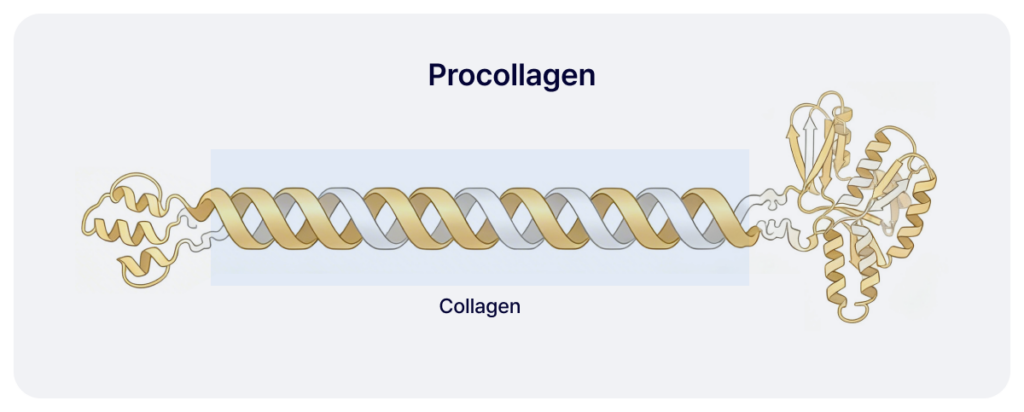
Collagen Triple Helix Structure
Collagen is synthesized within the cell in the form of procollagen, which is then converted into mature collagen through proteolytic cleavage.
Procollagen consists of three polypeptide α-chain that wind together to form a triple helix structure. This structure can exist as a homotrimer (composed of identical chains) or a heterotrimer (composed of different chains).
At the core of the triple helix domain, there is a characteristic repeating sequence:
Gly–X–Y motif
- Gly: glycine
- X / Y: Primarily proline or hydroxyproline
This repetitive structure is essential for the formation of a stable collagen triple helix. Subsequently, the N- and C-terminal propeptides are removed from the procollagen through proteolytic cleavage, resulting in the formation of mature collagen molecules.
Causes of Classical EDS (cEDS)
Classical EDS is caused by defects in Type V collagen.
Type V collagen typically exists as a heterotrimer structure, where a single heterotrimer is composed of two α1(V) chains and one α2(V) chain.
The genes encoding these $\alpha$-chains are as follows:
- α1(V): COL5A1
- α2(V): COL5A2
→ These play an essential role in collagen fibrillogenesis.
1. COL5A1 Variant Mechanism
Most COL5A1 variants cause haploinsufficiency.
Common variant types include:
- nonsense
- frameshift
These variants typically trigger nonsense-mediated mRNA decay (NMD).
Consequently, the production of normal Type V collagen is reduced by approximately 50%.
2. COL5A2 Variant Mechanism
Variants in COL5A2 are usually:
- missense
- in-frame indel
These variants disrupt collagen structure directly and often result in more severe phenotypes.
Vascular EDS
Vascular EDS is caused by pathogenic variants in COL3A1.
Type III collagen plays an important role in:
- collagen fibril assembly
- regulation of fibril diameter
The most common pathogenic variants include:
- glycine substitutions
- in-frame insertions/deletions
These variants cause:
- collagen misfolding
- failure of collagen secretion
- retention within the endoplasmic reticulum
This results in a dominant-negative effect.
Consequently, ECM integrity is compromised, leading to:
- arterial rupture
- organ rupture
which are hallmark complications of vascular EDS.
Type I Collagen–Related EDS
Type I collagen is the most abundant protein in the extracellular matrix (ECM).It has a heterotrimer structure, consisting of two α1(I) chains and one α2(I) chain.
Associated genes include:
- COL1A1
- COL1A2
While most variants cause osteogenesis imperfecta, some result in specific EDS subtypes. The primary pathological mechanism is the failure of normal collagen fibrillogenesis due to abnormalities in N-propeptide cleavage and the procollagen processing steps.
Arthrochalasia EDS (aEDS)
- Cause: Heterozygous
COL1A1/COL1A2variants (partial or total deletion of exon 6). - Loss of the N-propeptide cleavage site.
- Loss of cross-linking lysine, protease cleavage site, and initial triple-helix triplets.
- Failure to remove the N-terminal of Type I procollagen, leaving it partially attached.
- Abnormal procollagen processing occurs → Impaired collagen fibrillogenesis.
Key Features: Severe joint hypermobility and congenital hip dislocation.
Dermatosparaxis EDS (dEDS)
Cause: ADAMTS2 variants / Biallelic loss-of-function of ADAMTS2.
Mechanism:
Failure of procollagen N-cleavage.
Abnormal collagen fiber formation.
Key Features: Extreme skin fragility and severe bruising.
Diagnostic Clue: “Hieroglyphic” collagen fibrils are observed under electron microscopy.
Electron microscopy reveals characteristic hieroglyphic collagen fibrils.
Dermatosparaxis EDS (dEDS)
- Cause: ADAMTS2 mutation / Biallelic loss of function (LoF) in ADAMTS2.
- Mechanism: Failure of procollagen N-cleavage leads to abnormal collagen fibril formation.
- Characteristics: Extreme skin fragility and severe bruising.
- Diagnosis: “Hieroglyphic” collagen fibril morphology is observed under electron microscopy.
Type I Collagen Related Mechanisms
Cardiac Valvular EDS (cvEDS)
- Mechanism: Biallelic COL1A2 LoF mutation.
- Pathology: mRNA instability due to Nonsense-Mediated Decay (NMD) → Deficiency of $pro\alpha2(I)$ chains → Formation of $\alpha1(I)$ homotrimer collagen.
- Clinical Feature: Severe polyvalvular heart disease.
Contrasting Mechanism: Osteogenesis Imperfecta (OI)
- Mechanism: Biallelic COL1A2 LoF where mRNA remains stable.
- Pathology: Production of unstable proteins → Activation of the Unfolded Protein Response (UPR/ER stress).
- Result: Mild-to-moderate Osteogenesis Imperfecta.
- Comparison: This highlights the pathological difference between ECM defects vs. ER stress-based mechanisms.
COL1A1 Arg → Cys Mutation
- Mechanism: Heterozygous substitution of Arginine with Cysteine in COL1A1.
- Pathology: Cysteine is introduced into the triple helix → Formation of abnormal disulfide-bond dimers → Localized collagen structural instability and altered ECM interactions.
- Phenotype: Risk of arterial rupture (similar to vEDS) ± skin/joint symptoms (similar to cEDS).
- Spectrum: Some mutations mimic cEDS (without COL5 mutations), while others show EDS/OI overlap (joint hypermobility + mild bone fragility).
Collagen Crosslinking & Folding Defects: kEDS
EDS can occur not only during fibrillogenesis but also during the folding and cross-linking stages. These typically present as Kyphoscoliotic EDS.
Kyphoscoliotic EDS (kEDS)
PLOD1 Deficiency: Deficiency of lysyl hydroxylase → Impaired collagen crosslinking.
- ~30% of mutations involve duplication of exons 10–16 via Alu-mediated recombination.
Features: Progressive scoliosis, vascular fragility.
FKBP14 Deficiency: ER chaperone protein defect → Impaired collagen folding.
Result: ER collagen accumulation and abnormal fibril formation.
Note: A founder frameshift variant accounts for most alleles.
ECM Structural Protein Defects
Classical-like EDS (clEDS)
- Cause: Biallelic LoF mutations in TNXB (Tenascin-X deficiency).
- Result: Impaired collagen fibril organization.
- Symptoms: Skin hyperextensibility (without atrophic scarring), easy bruising, joint hypermobility ± muscle weakness.
- Heterozygotes: Reduced TNX levels; some show joint hypermobility (rarely linked to hEDS).
Myopathic EDS (mEDS)
- Cause: Heterozygous or biallelic mutations in COL12A1 (Type XII collagen).
- Features: EDS combined with myopathy. Biallelic mutations cause severe congenital forms, while heterozygous mutations result in milder phenotypes.
Glycosaminoglycan (GAG) Synthesis Defects
Proteoglycans maintain ECM structure, regulate collagen spacing, and modulate cell signaling. They consist of a core protein and GAG chains (HS, CS, DS).
Spondylodysplastic EDS (spEDS)
- Cause: Mutations in B4GALT7 or B3GALT6.
- Mechanism: Defects in the galactosylation of the tetrasaccharide linker.
- Result: Inability to add sugars to core proteins; reduction/shortening of Heparan Sulfate (HS) and Chondroitin Sulfate (CS); loss of Dermatan Sulfate (DS) from decorin.
- Features: Skeletal dysplasia, hypotonia.
Musculocontractural EDS (mcEDS)
- Cause: Biallelic mutations in CHST14 or DSE.
- Mechanism: Impaired DS synthesis; DS is replaced by CS.
- Result: Dysfunction of decorin leads to disrupted ECM structure and collagen organization.
- Features: Congenital contractures, craniofacial anomalies.
Other Intracellular & Complement Molecule Defects
spEDS (SLC39A13-related)
- Mechanism: Loss of ZIP13 zinc transporter function → Decreased zinc influx into the cytoplasm.
- Result: Reduced collagen lysyl/prolyl hydroxylation → ECM crosslinking defects.
- Proposed Pathogenesis: Zn/Fe imbalance, ER stress, and altered BMP/TGF-$\beta$ signaling.
Brittle Cornea Syndrome (BCS)
- Cause: Biallelic ZNF469 or PRDM5 mutations.
- Mechanism: PRDM5 is a transcription factor regulating ECM genes.
- Features: Extremely thin and fragile corneas, high risk of corneal rupture.
Periodontal EDS (pEDS)
Cause 1: Heterozygous C1R or C1S mutations (Classical complement pathway).
Mechanism: GoF (Gain of Function) activation of C1 protease → Increased local complement activity.
Cause 2: Biallelic AEBP1 mutations (Loss of ACLP, a collagen-binding protein).
Mechanism: Impaired Collagen I polymerization and altered TGF-βWnt signaling.
Features: Aggressive periodontitis, early tooth loss, skin hyperextensibility.
EDS Diagnosis & Challenges
Common Clinical Features
- Generalized joint hypermobility
- Skin hyperextensibility and tissue fragility
- Easy bruising and abnormal wound healing
Why Diagnosis is Difficult
- Subtlety in Children: Tissue fragility may be mistaken for the “normal” range.
- Physical Masking: Subcutaneous fat can mask skin hyperextensibility in infants.
- Delayed Presentation: Brusing and skin injuries often only become apparent after the child starts walking; severe complications like vascular rupture (vEDS) typically emerge after adolescence.
References
Malfait, F. et al. The Ehlers–Danlos syndromes. Nat. Rev. Dis. Primers 6, 64 (2020).
Malfait, F. et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 175, 8–26 (2017).
Blackburn, P. R. et al. Bi-allelic alterations in AEBP1 lead to defective collagen assembly and connective tissue structure resulting in a variant of Ehlers–Danlos syndrome. Am. J. Hum. Genet. 102, 696–705 (2018).
Wenstrup, R. J. et al. What mouse mutants teach us about extracellular matrix function. Annu. Rev. Cell Dev. Biol. 22, 591–621 (2006).
Gao, L., Orth, P., Cucchiarini, M. & Madry, H. Effects of solid acellular type-I/III collagen biomaterials on chondrogenesis of mesenchymal stem cells. Expert Rev. Med. Devices (2017).
Zhang, X. et al. Procollagen-lysine 2-oxoglutarate 5-dioxygenase 2 promotes collagen cross-linking and ECM stiffening to induce liver fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 1870, 167205 (2024).
Lloyd, S. M. & He, Y. Exploring extracellular matrix crosslinking as a therapeutic approach to fibrosis. Cells 13, 438 (2024).
doi:10.3390/cells13050438
Ishikawa, Y. & Bächinger, H. P. A substrate preference for the rough endoplasmic reticulum resident protein FKBP22 during collagen biosynthesis. J. Biol. Chem. 289, 18189–18201 (2014).
Lethias, C. et al.
A model of tenascin-X integration within the collagenous network.
FEBS Lett. 580, 6281–6285 (2006)
Nguyen, M. & Panitch, A. Proteoglycans and proteoglycan mimetics for tissue engineering. Am. J. Physiol. Cell Physiol. 322, C754–C761 (2022).
Ruiz Martínez, M. A., Peralta Galisteo, S., Castán, H. & Morales Hernández, M. E. Role of proteoglycans on skin ageing: a review. Int. J. Cosmet. Sci. 42, 529–535 (2020).
Green, C. et al. Classical-like Ehlers–Danlos syndrome: a clinical description of 20 newly identified individuals with evidence of tissue fragility. Genet. Med. 22, 1576–1582 (2020).
Abdalla, E. M. et al. Kyphoscoliotic type of Ehlers–Danlos syndrome (EDS VIA) in six Egyptian patients presenting with a homogeneous clinical phenotype. Eur. J. Pediatr. 174, 105–112 (2015).
Chen, Y. et al. Role of genetics in amyotrophic lateral sclerosis: a large cohort study in Chinese mainland population. J. Med. Genet. 59, 840–849 (2022).
Get exclusive rare disease updates
from 3billion.

3billion Inc.
3billion is dedicated to creating a world where patients with rare diseases are not neglected in diagnosis and treatment.