Episode 6 – Cornelia de Lange Syndrome (CdLS): Beyond the Variant
📌 Series Introduction
In an era where AI-based variant interpretation tools have become widely adopted, this series focuses on what human interpreters must still understand and decide after automated prioritization.
Each episode centers on a specific disease group, examining genetic mechanisms and clinical spectra together, with the goal of moving beyond simply “finding variants” toward explaining and interpreting them in a clinically meaningful way.
Cornelia de Lange syndrome (CdLS)
Key Takeaway
CdLS is a condition suspected from phenotype, confirmed through variant interpretation, and only becomes scalable when that interpretation is structured and accumulated.
What is CdLS?
Cornelia de Lange syndrome (CdLS) is a rare genetic disorder that has been recognized for over a century.
It was first reported in 1849, and later clinically characterized by Cornelia de Lange in the 1930s.
In 2004, the discovery of the NIPBL gene provided the first molecular explanation of the disease.
👉 In other words, CdLS is a condition that was defined clinically first, and only later understood genetically.
What does it look like clinically?
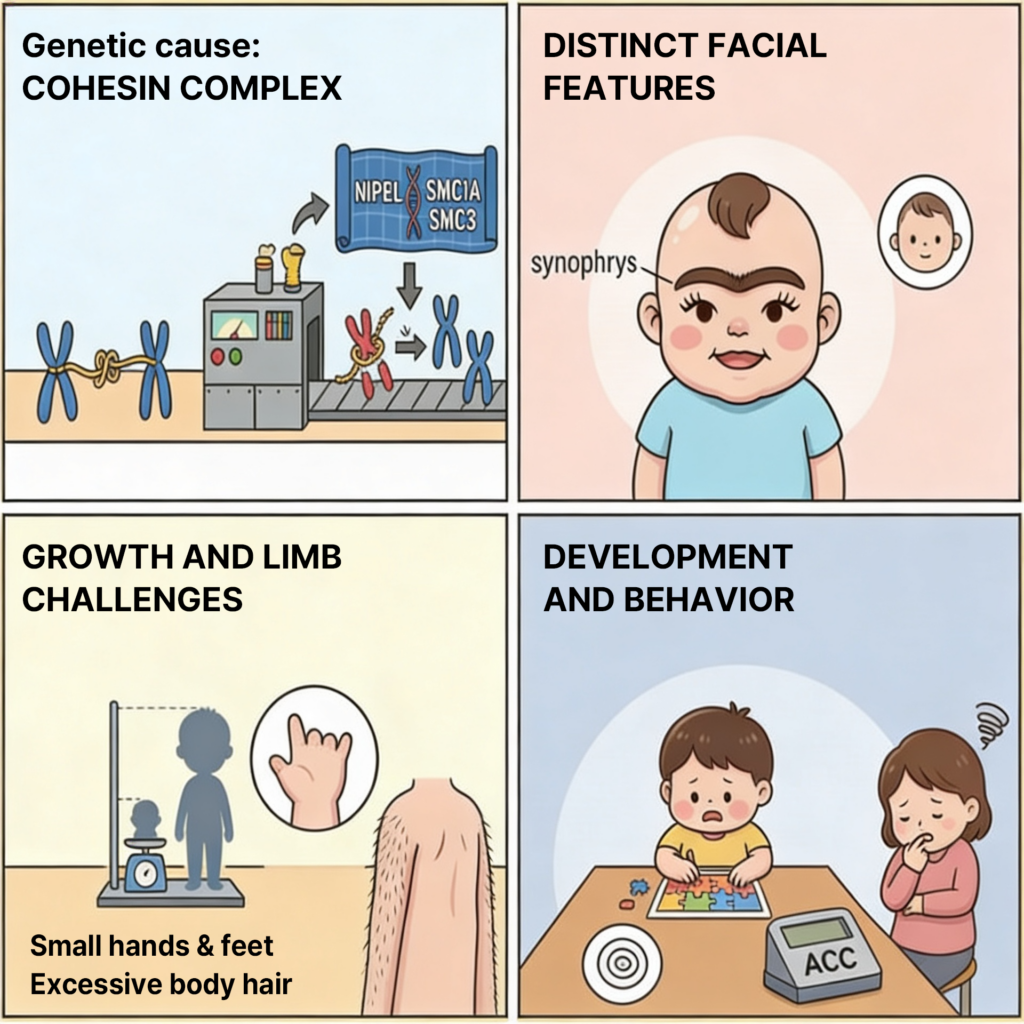
CdLS is a classic syndromic disorder affecting multiple systems.
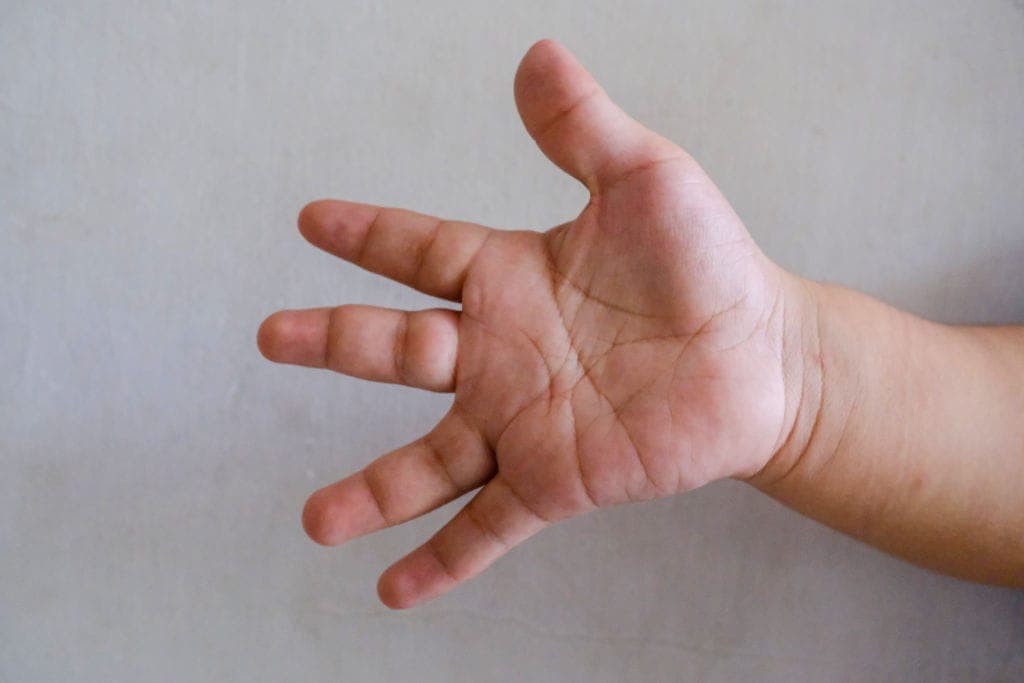
Key craniofacial features include synophrys, hirsutism, and anteverted nares, which often serve as the first clues.
These are commonly accompanied by: growth delay, neurodevelopmental disorders, self-injurious behavior
In addition, patients may present with: upper limb anomalies, GERD, congenital heart defects, hearing loss
👉 In practice, CdLS can be understood across three axes: face + growth/development + limb anomalies
But is that enough for diagnosis?
Historically, diagnosis relied heavily on recognizable clinical features.
If these features were present, one might conclude: “this looks like CdLS.”
However, in real clinical settings, diagnosis is often not that straightforward.
- Phenotypic variability is high
- Many cases are atypical
- Some resemble entirely different syndromes
As our genetic understanding evolved, it became clear that:
❗ CdLS is not a condition that can be confirmed by appearance alone
❗ Diagnosis is only complete when the underlying variant is identified
Why is CdLS so variable?
CdLS is now understood as a cohesinopathy, a spectrum disorder rather than a single defined entity.
- The same gene can produce different phenotypes
- Different genes can produce similar phenotypes
This led to the concepts of:
- Classic CdLS
- Non-classic CdLS
- CdLS-like phenotypes
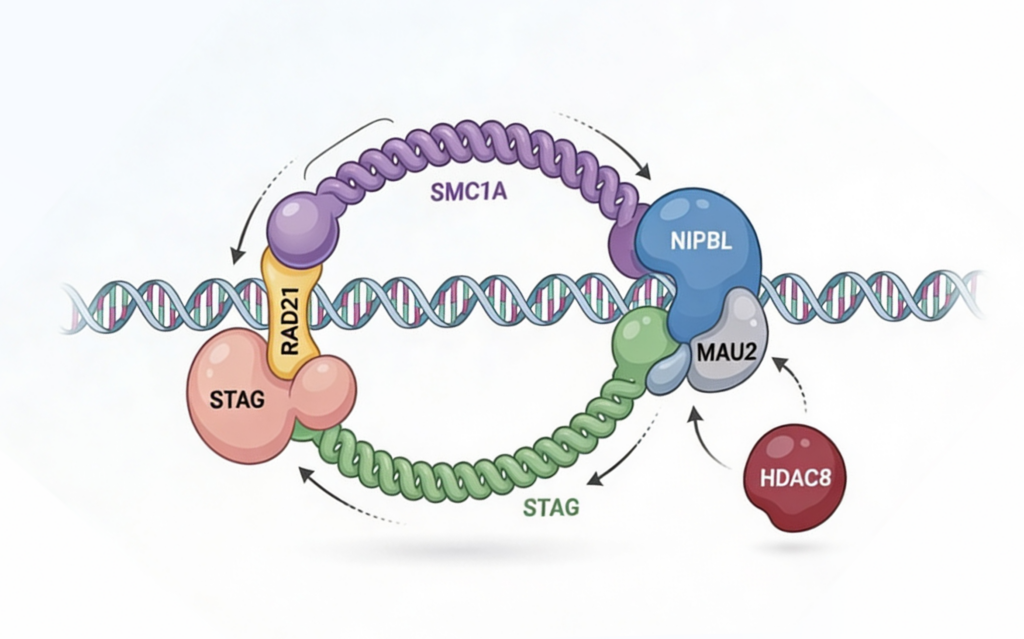
The core mechanism: transcriptional dysregulation
The key to understanding CdLS is not cohesin structure itself, but its role in transcription regulation.
- alterations in chromatin structure
- disruption of enhancer–promoter interactions
- global changes in gene expression
👉 The result is not a single disrupted pathway, but a subtle, system-wide dysregulation of gene expression
This explains why phenotypes vary so widely.
Looking at genes helps — but only to a point
CdLS is associated with multiple genes, each showing certain trends.
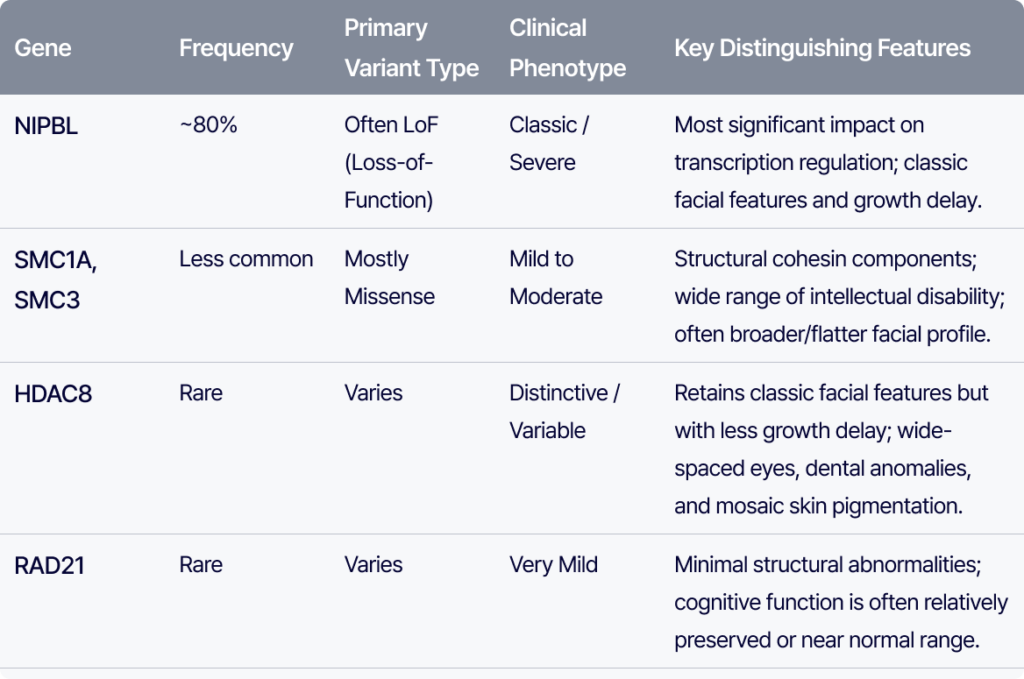
NIPBL (~80%) is the most common cause. Loss-of-function variants are frequent, and due to its central role in transcription regulation, it is often associated with classic and more severe phenotypes.
In contrast, SMC1A and SMC3 encode structural components of the cohesin complex. These are typically associated with missense variants and generally present with milder phenotypes, though intellectual disability can vary.
HDAC8 cases often retain typical facial features, but growth delay is less pronounced. Clues include wide-spaced eyes, a broad nose, dental anomalies, and mosaic skin pigmentation.
RAD21 is associated with minimal structural abnormalities, and cognitive function is often relatively preserved.
👉 In summary:
- NIPBL → more severe
- SMC group → mild–moderate
- RAD21 → often mild
However, These are not strict rules. They are tendencies, not boundaries.
Where interpretation becomes difficult
In practice, many cases fall into a gray zone.
- A relevant variant is identified
- But the phenotype is not fully convincing
- Confidence remains low
Or, the same data leads to different conclusions across teams
This becomes especially problematic in conditions like CdLS, where interpretation — not detection — is the bottleneck
The key question
Ultimately, CdLS interpretation comes down to one question:
👉 “Does this variant truly explain this patient?”
Answering this requires more than annotation.
It requires structured reasoning:
- linking phenotype to gene
- understanding functional impact
- comparing with differential diagnoses
Where many teams struggle
This is often where workflows break down.
- Interpretations are made, but not recorded
- The same variant is reinterpreted repeatedly
- Decision criteria vary across individuals
👉 As a result, interpretation becomes dependent on individual experience
rather than a reproducible system.
Interpreter Tips
- Phenotype is only the starting point
- Do not dismiss atypical presentations too quickly
- For NIPBL, be able to explain why the phenotype is severe
- Always consider CdLS-like conditions
👉 And most importantly:
Interpretation must be documented to be reusable
One-line summary
👉 CdLS is a condition suspected from phenotype, confirmed through variant interpretation, and only becomes scalable when that interpretation is structured and accumulated.
What this means in practice
As discussed above, in conditions like CdLS, the challenge is not simply finding variants — it is how interpretation is structured, retained, and reused
💡 If you’ve ever thought: “Is there a more systematic way to approach cases like this?”
GEBRA is designed to support exactly that layer of interpretation:
- connecting phenotype and variant
- capturing interpretation rationale
- enabling a repeatable interpretation workflow
with the goal of making clinical interpretation faster, more consistent, and scalable
References
- Parenti I, Kaiser FJ. Cornelia de Lange syndrome as paradigm of chromatinopathies. Front Neurosci. 2021;15:774950. doi:10.3389/fnins.2021.774950
- Deardorff MA, Noon SE, Krantz ID. Cornelia de Lange syndrome. In: Adam MP, Bick S, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2026 [updated 2020 Oct 15]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1104/
Sarogni P, Pallotta MM, Musio A. Cornelia de Lange syndrome: from molecular diagnosis to therapeutic approach. J Med Genet. 2020;57(5):289–295. doi:10.1136/jmedgenet-2019-106277 - Minor A, Shinawi M, Hogue JS, Vineyard M, Hamlin DR, Tan C, Donato K, Wysinger L, Botes S, Das S, Del Gaudio D. Two novel RAD21 mutations in patients with mild Cornelia de Lange syndrome-like presentation and report of the first familial case. Gene. 2014;537(2):279–284. doi:10.1016/j.gene.2013.12.045
- Santen GW, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M, Kant SG, Snoeck IN, Peeters EA, Hilhorst-Hofstee Y, Wessels MW, den Hollander NS, Ruivenkamp CA, van Ommen GJ, Breuning MH, den Dunnen JT, van Haeringen A, Kriek M. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet. 2012;44(4):379–380. doi:10.1038/ng.2217
- Raible SE, Mehta D, Bettale C, Fiordaliso S, Kaur M, Medne L, Rio M, Haan E, White SM, Cusmano-Ozog K, Nishi E, Guo Y, Wu H, Shi X, Zhao Q, Zhang X, Lei Q, Lu A, He X, Okamoto N, Miyake N, Piccione J, Allen J, Matsumoto N, Pipan M, Krantz ID, Izumi K. Clinical and molecular spectrum of CHOPS syndrome. Am J Med Genet A. 2019;179(7):1126–1138. doi:10.1002/ajmg.a.61174
Get exclusive rare disease updates
from 3billion.

3billion Inc.
3billion is dedicated to creating a world where patients with rare diseases are not neglected in diagnosis and treatment.