Episode 4 Bardet-Biedl Syndrome (BBS): Beyond the Variant
📌 Series Introduction
In an era where AI-based variant interpretation tools have become widely adopted, this series focuses on what human interpreters must still understand and decide after automated prioritization.
Each episode centers on a specific disease group, examining genetic mechanisms and clinical spectra together, with the goal of moving beyond simply “finding variants” toward explaining and interpreting them in a clinically meaningful way.
BBS is a condition with such a wide range of symptoms that it is difficult to consider it as a single disease.
Why does BBS present with such diverse manifestations?
1. What is Bardet-Biedl Syndrome?
Bardet-Biedl syndrome (BBS) is a rare genetic disorder that affects multiple organs simultaneously.
The name of the disease originates from two physicians:
- Georges Bardet (France)
- Arthur Biedl (Hungary)
They independently described patients with similar clinical features, which led to the naming of the condition.
The prevalence of BBS is as follows:
- General population: approximately 1/140,000–1/160,000 (North America, Europe)
- Middle East: approximately 1/13,500 (Kuwait)
The higher prevalence in the Middle East is largely due to autosomal recessive inheritance combined with consanguinity.
Based on internal data from 3billion, a total of 105 patients have been diagnosed with Bardet-Biedl syndrome to date—many of whom had previously experienced long diagnostic journeys across multiple specialties without a clear cause.
2. The Key Concept: A Cilia Disorder (Ciliopathy)
To understand BBS, we must first understand cilia.
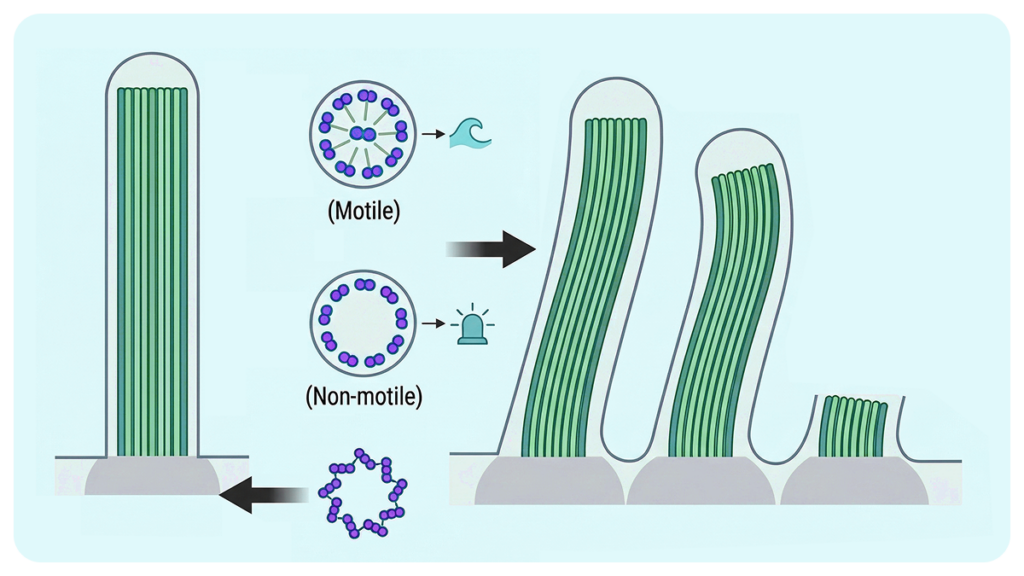
What are cilia?
They are small structures located on the surface of eukaryotic cells, broadly classified into two types:
- Primary cilia (non-motile)
→ Function as sensory organelles that detect external signals - Motile cilia
→ Involved in cell movement and transport of substances (e.g., mucus, sperm, cerebrospinal fluid)
These two types of cilia have distinct internal structures.
BBS is closely associated with dysfunction of primary cilia.
3. Core Mechanism: What Goes Wrong?
Primary cilia are not just structural components—they function as signaling platforms.
They detect external stimuli such as chemosensation, mechanosensation, and hormonal signals, and transmit them into the cell.
Within this system, the following mechanisms operate:
- Kinesin / Dynein (motor proteins)
- IFT (intraflagellar transport)
In other words, there is a system that transports proteins to the tip of the cilium.
The core of BBS can be summarized in one line:
- Disruption of protein transport within cilia
- Abnormal assembly and regulation of the BBSome complex
- Defects in ciliogenesis
- Impaired protein trafficking and turnover
As a result, signaling pathways collapse, leading to dysfunction across multiple organs.
4. Major Clinical Features
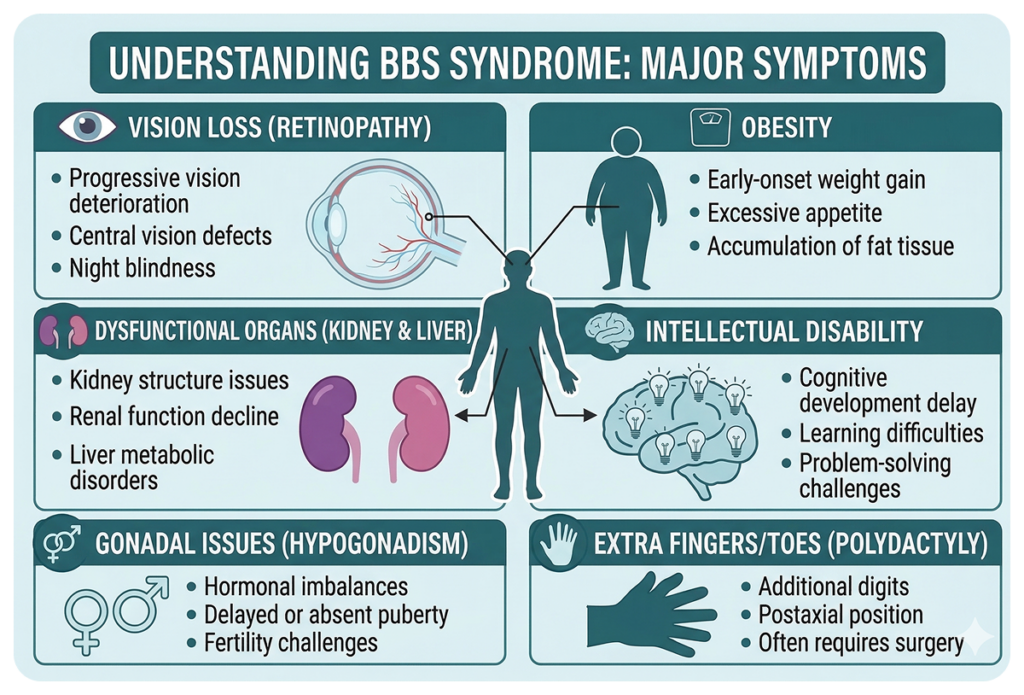
The representative symptoms of BBS include:
🔴 Major symptoms
- Retinopathy
- Obesity
- Polydactyly
- Kidney dysfunction
- Hypogonadism
- Developmental delay / Intellectual disability
A wide phenotypic spectrum has been reported, and the frequency of each symptom varies.
Among these, the combination of retinopathy + obesity is a particularly important clinical clue.
However, there are important considerations in diagnosis:
Unlike diseases such as CMT, where clinical features are relatively well stratified depending on the causative gene, BBS shows broadly similar clinical manifestations despite a wide range of associated genetic variants.
Symptoms typically begin in childhood, but the onset of each feature varies and is often progressive. As a result, early diagnosis can be difficult, and in some patients, the clinical picture becomes clear only in their 20s.
Furthermore, major features such as retinal abnormalities and obesity do not show 100% penetrance, which can lead to delayed or missed diagnoses.
5. Why Are the Symptoms So Diverse?
There is one key reason:
Cilia are present in almost all organs.
Including:
- Retina
- Kidney
- Brain
- Gonads
- Adipose tissue
Therefore, a single defect in cilia leads to a multi-system disorder.
6. Pathophysiology by Symptom (Key Understanding)
① Retinopathy (vision loss)
Cilia are present in rod and cone cells of the retina
→ Ciliary dysfunction leads to impaired cellular function → vision decline
② Kidney dysfunction
Cilia are also present in renal tubules
→ Loss of function leads to renal abnormalities (mechanism not fully understood)
Polycystic kidney:
Ciliary dysfunction → disrupted calcium signaling → loss of proliferation control → cyst formation
③ Polydactyly
Abnormal Sonic hedgehog (SHH) signaling
→ Failure in limb axis determination
→ Occurs during development
④ Hypogonadism
Dysfunction of GnRH neurons
→ Impaired kisspeptin signaling → decreased GnRH secretion → hypogonadism
(mainly based on animal models)
⑤ Intellectual disability
- Wnt signaling: required for organ development, axonal guidance, neuronal migration, synaptogenesis
- Hedgehog signaling: important for neural tube patterning and neural stem cell development
Ciliary dysfunction disrupts Wnt / Hedgehog signaling
→ impaired neural connectivity
→ abnormal neuronal networks
⑥ Obesity (most intriguing aspect)
Causes:
- Hypothalamic dysfunction
- Leptin receptor dysfunction → failure to sense satiety
- Abnormal adipocyte differentiation
→ due to disrupted Wnt / SHH signaling - Abnormal energy metabolism
→ due to non-canonical SHH signaling
→ reduced glucose uptake and thermogenesis
Result: hyperphagia + disrupted energy balance
7. Genetic Background
- Inheritance: Autosomal recessive (AR)
- Number of genes: over 26
More specifically:
- BBSome complex components: BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9, BBIP10
- Chaperone-like proteins: BBS6, BBS10, BBS12
- Ciliogenesis (cilia formation): BBS13, BBS14, BBS15
- Protein transport / turnover:
– Intraflagellar transport: BBS3, BBS17
– IFT-B complex: BBS19, BBS20
– Protein ubiquitination: BBS11
These genes are involved in diverse biological pathways linked to disease mechanisms.
8. Summary (Key Take-home)
Bardet-Biedl syndrome can be summarized as:
✔ A ciliopathy
✔ Core cause: defects in ciliary transport + ciliogenesis
✔ Major symptoms:
- Retinopathy
- Obesity
- Polydactyly
- Kidney dysfunction
- Hypogonadism
- Intellectual disability
✔ Why it is systemic:
→ because cilia are present in almost all organs
What matters starts from here.
Symptoms that appear completely unrelated when viewed separately can only reveal the diagnosis of BBS when they are connected through a single underlying mechanism.
AI can rapidly narrow down candidate variants among thousands.
But our role is not simply to find variants.
The essence of interpretation lies in how convincingly we can explain the patient through that variant.
References
Forsythe, E. & Beales, P. L. Bardet-Biedl syndrome. Eur. J. Hum. Genet. 21, 8–13 (2013).
Haws, R. M., Joshi, A., Gigante, M. & Spira, M. Assessing the impact of setmelanotide on obesity and hyperphagia in Bardet-Biedl syndrome: a comprehensive review. Orphanet J. Rare Dis. 17, 148 (2022).
Melluso, A., Secondulfo, F., Capolongo, G., Capasso, G. & Zacchia, M. Bardet-Biedl syndrome: current perspectives and clinical outlook. Ther. Clin. Risk Manag. 19, 115–132 (2023).
Reiter, J. F. & Leroux, M. R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 18, 533–547 (2017).
Restaino, S., Pagliuca, M., Sperduti, I., Scambia, G. & Fanfani, F. ERGO (ERGonomics in the operating room) study: a cross-sectional international online survey. Surgery 175, 1045–1052 (2024).
Waschke, J. The desmosome: a target in pemphigus vulgaris and arrhythmogenic cardiomyopathy. Front. Cell Dev. Biol. 8, 608241 (2020).
Wilson, N. R., Olm-Shipman, A. J., Acevedo, D. S., Palaniyandi, K., Hall, E. G., Kosa, E. & Saadi, I. SPECC1L deficiency results in adherens junction defects and orofacial clefting. Front. Cell Dev. Biol. 6, 6 (2018).
Get exclusive rare disease updates
from 3billion.

3billion Inc.
3billion is dedicated to creating a world where patients with rare diseases are not neglected in diagnosis and treatment.