Episode 8: Lysosomal Storage Disorders (LSD) — Beyond Variant Interpretation
Key Takeaways
- Systemic Over Individual: Lysosomal Storage Disorders (LSDs) are not single-organ diseases; they are systemic conditions caused by disruptions in the intracellular degradation system.
- Pattern Over Nomenclature: In LSD interpretation, connecting the phenotype and the pattern of accumulated substrates is far more critical than simply guessing a specific disease name.
- Structured Interpretation Framework: Approaching cases in a structured sequence—Phenotype → Affected Organs → Accumulated Substrates → Biochemical Pathway → Candidate Genes—renders the interpretation workflow significantly more robust.
- Biomarkers as Anchors: Biomarkers and enzyme activity assays serve as definitive functional evidence that guides the direction of variant interpretation.
- The Causality Chain: Successful LSD analysis requires unifying the patient’s phenotype, biomarkers, enzyme function, and underlying disease mechanisms into a single, cohesive causality chain.
📌 Series Introduction
This series is authored by clinical genomics experts at 3billion. In an era where AI-driven variant prioritization tools have become ubiquitous, we focus on what interpreters must critically evaluate and decide after automated prioritization is complete.
In each episode, we focus on a specific disease group, exploring both the underlying genetic mechanisms and the clinical spectrum. Our goal is to share insights that move beyond merely “finding” variants to truly “explaining and interpreting” them.
Why is “Connecting the Clues” Critical in LSD Interpretation?
Lysosomal Storage Disorders (LSDs) constitute a complex category of rare genetic conditions that present with a broad and highly variable spectrum of clinical phenotypes.
While disorders like Gaucher disease, Fabry disease, Niemann-Pick disease, Tay-Sachs disease, Mucopolysaccharidosis (MPS), and Pompe disease are well-documented, actual clinical cases rarely present exactly as textbooks describe.
A patient might present initially with isolated hepatosplenomegaly, or perhaps with a predominantly neurodegenerative course. In other scenarios, clinicians are faced with a scattered constellation of multi-organ symptoms: cardiomyopathy, progressive renal failure, skeletal dysplasia, proximal muscle weakness, neuropathic pain, or angiokeratomas.
Consequently, the fundamental question in LSD interpretation is not simply:
- “Does this patient have Fabry disease?”
- “Is this Gaucher disease?”
Instead, the foundational question must be:
Can this patient’s multi-systemic manifestations be explained by a singular, underlying substrate storage pathology?
Why do LSDs Manifest Systemically?
Lysosomes function as the primary recycling and degradation centers of the cell. When a specific lysosomal enzyme is deficient, a trafficking protein malfunctions, or an enzyme fails to target the lysosome properly, the substrates destined for degradation progressively accumulate within the organelle.
Over time, this progressive cellular crowding disrupts vital cellular processes, triggering a cascade of tissue damage and organ dysfunction.
For the genomic interpreter, this underscores a critical reality: LSDs are not localized organ issues. They represent systemic phenotypic consequences born from a failure in cellular clearance.
Therefore, when a patient presents with overlapping symptoms across the nervous system, heart, liver, spleen, skeleton, muscles, or kidneys, the interpreter must pivot away from an isolated, symptom-centric viewpoint. Instead, they must approach the data with a high index of suspicion for an underlying storage defect.
The Core of LSD Analysis: “What is Accumulating?”
LSDs involve numerous distinct conditions and a massive array of associated genes. From an interpretive standpoint, trying to memorize every single clinical entity is inefficient. A more powerful approach is to stratify your analysis based on the accumulated substrate.
- Sphingolipid Accumulation: Points directly toward sphingolipidoses, such as Gaucher disease, Fabry disease, or Tay-Sachs disease.
- Glycosaminoglycan (GAG) Accumulation: Signals the Mucopolysaccharidosis (MPS) spectrum.
- Glycogen Accumulation: Establishes Pompe disease as a primary differential candidate.
- Cholesterol/Lipid Trafficking Defects: Suggests a transporter defect, such as Niemann-Pick disease type C.
By leveraging this biochemical reality, we utilize the following structured framework:
🧬 LSD Interpretation Framework
Phenotype → Affected Organs → Accumulated Substrates → Related Pathway → Candidate Genes
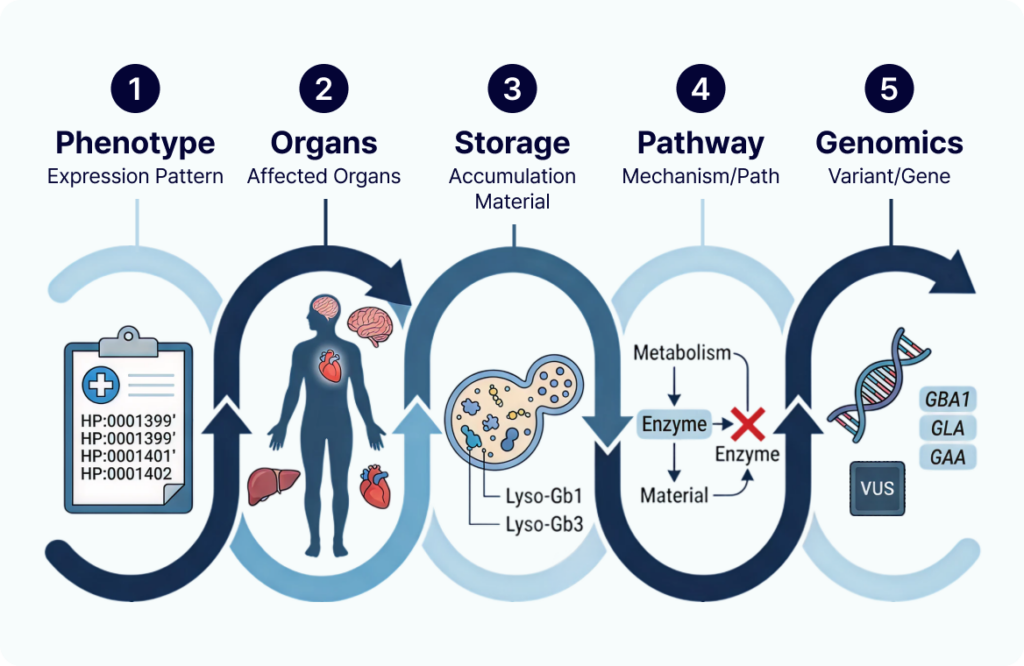
Adhering to this pipeline allows you to systematically narrow down candidate genes in a clinically meaningful way, far outperforming standard, isolated variant filtering.
Reading Gaucher, Fabry, and Pompe as “Patterns”
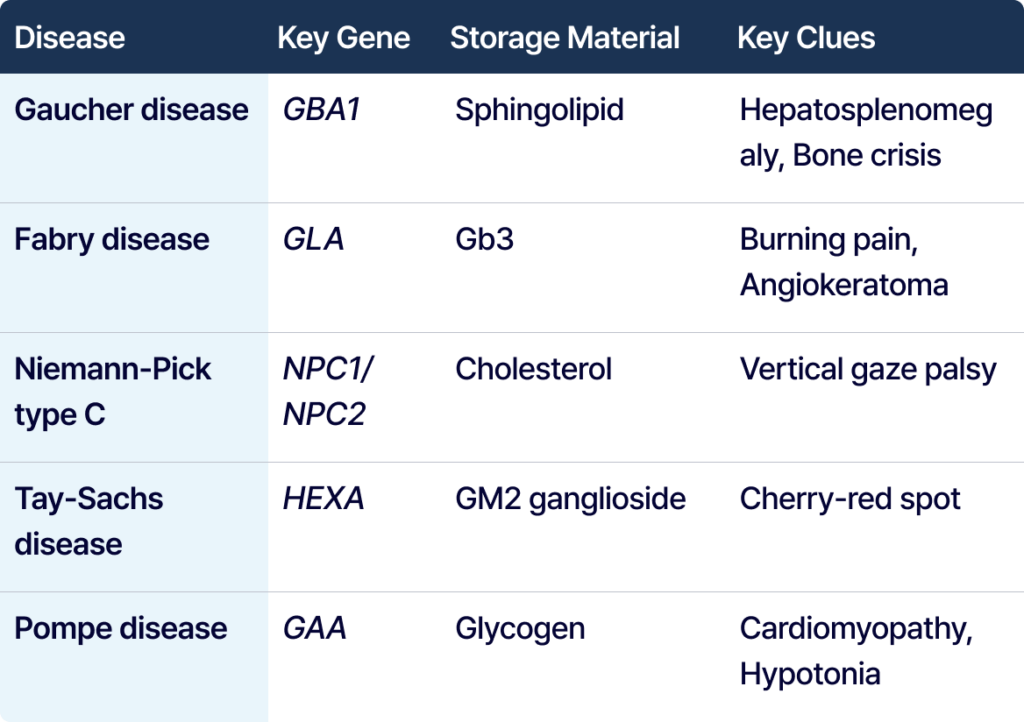
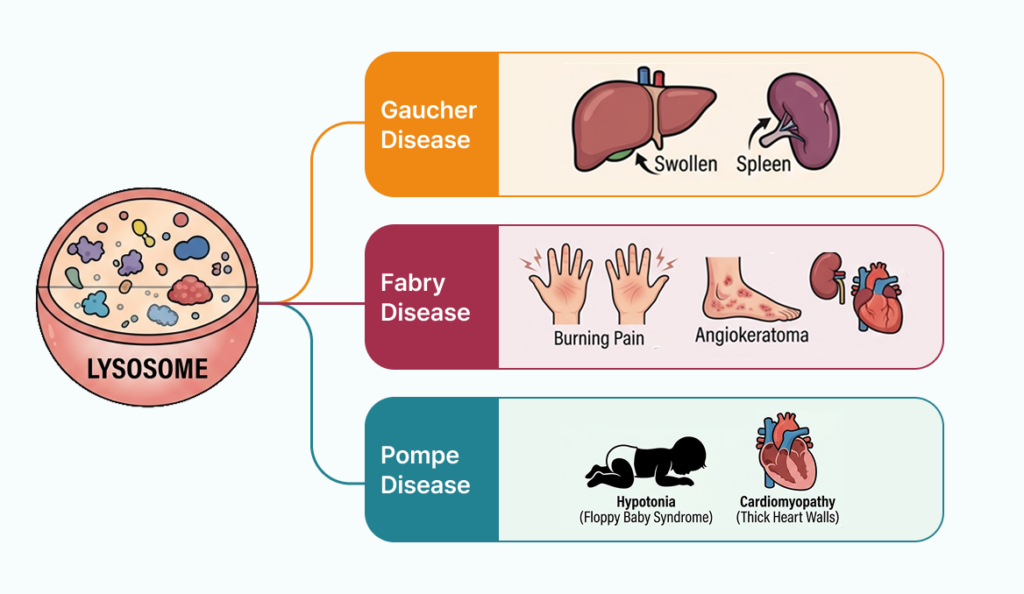
Gaucher disease is linked to the GBA1 gene. Crucial indicators include hepatosplenomegaly, anemia, thrombocytopenia, bone pain, bone crises, and osteonecrosis. GBA1 should be carefully considered, especially when hepatosplenomegaly is accompanied by hematologic abnormalities and bone involvement.
Fabry disease is an X-linked condition linked to the GLA gene. Major indicators include burning pain in the extremities, angiokeratoma, hypohidrosis, and renal/cardiac involvement, along with early-onset stroke risk. Because it can initially manifest as adult-onset cardiomyopathy or renal disease, interpretation alongside biomarkers like Lyso-Gb3 is critical.
Niemann-Pick disease must be differentiated into type A/B and type C. Type A/B involves SMPD1-associated acid sphingomyelinase deficiency, while type C is a cholesterol trafficking defect due to NPC1 or NPC2 mutations. For type C, a presentation combining vertical gaze palsy, ataxia, cognitive decline, and hepatosplenomegaly is highly indicative.
Tay-Sachs disease is linked to the HEXA gene. Crucial indicators include infantile neurodegeneration, developmental regression, hyperacusis, and the presence of a cherry-red spot.
MPS is a cluster of GAG storage diseases. Diagnosis should not rely only on coarse facial features; a systemic pattern must be assessed, including skeletal abnormalities, short stature, joint contractures, organomegaly, airway issues, hearing loss, and developmental delay.
Pompe disease is a lysosomal glycogen storage disorder linked to the GAA gene. Crucial signs in infantile-onset include hypotonia, feeding difficulty, and hypertrophic cardiomyopathy. For late-onset, the core clues are proximal muscle weakness, respiratory muscle weakness, and exercise intolerance.
Ultimately, genomic analysis in LSDs excels when you determine whether the patient’s entire clinical phenotype aligns with a specific accumulated substrate and its corresponding pathophysiological mechanism.
The Role of Biomarkers in Resolving VUS
LSD cases frequently cannot be resolved solely through genomic sequencing data. It is highly common to encounter Variants of Uncertain Significance (VUS) or atypical, mild phenotypes. In these deadlocks, biochemical biomarkers serve as the ultimate arbiters.
Key LSD Biomarkers:
- Fabry disease → Lyso-Gb3
- Gaucher disease → Lyso-Gb1
- MPS → Urinary GAG
- Pompe disease → enzyme activity assay
As an interpreter, genomic variants, phenotypes, biomarkers, and enzyme assays cannot be evaluated in silos. They must be forged into a single causality chain:
- Does the identified genetic variant robustly explain the reduction in enzyme function?
- Does this enzymatic deficiency account for the specific substrate accumulation?
- Does the accumulation of this substrate match the patient’s multi-organ clinical pattern?
When these three dimensions lock into place, the diagnostic validity of your clinical report becomes bulletproof.
Why NGS Alone Falls Short in LSD Diagnosis
Next-Generation Sequencing (NGS) modalities—including targeted panels, whole-exome sequencing (WES), and whole-genome sequencing (WGS)—have revolutionized rare disease diagnostics. They are indispensable for uncovering atypical phenotypes, late-onset conditions, or overlapping clinical presentations.
However, an NGS pipeline merely identifies candidate variants; it does not render a final clinical diagnosis.
The true objective of LSD interpretation extends beyond finding a variant. You must definitively establish:
- If the variant aligns with the known disease mechanism.
- If it fully explains the patient’s clinical phenotype.
- Whether it stands congruent with biochemical biomarker profiles or functional enzyme assays.
Weaving these disparate data points into an unassailable diagnostic hypothesis is the indispensable role of the clinical genomics interpreter.
How GEBRA Empowers the LSD Interpretation Workflow
GEBRA is an advanced rare disease interpretation platform engineered to seamlessly bridge the gap between variants, genes, phenotypes, and clinical evidence within a single, integrated interface.
For complex disease groups like LSDs—where phenotypes are scattered across multiple organ systems and the integration of biochemical data is paramount—rudimentary variant filtering is insufficient.
GEBRA enables interpreters to dynamically investigate critical clinical questions:
- Is this specific variant mechanically consistent with the disease pathology?
- How precisely do the patient’s HPO terms map to the disease phenotype?
- Is the gene-disease relationship supported by robust, up-to-date evidence?
- Is there a compelling clinical context that justifies reporting a VUS?
- What complementary biomarker or functional evidence must be reconciled?
By consolidating these workflows, GEBRA enables clinical teams to evaluate complex genomic data with unprecedented speed, consistency, and diagnostic accuracy.
Conclusion
Interpreting Lysosomal Storage Disorders is not an exercise in matching a patient to a static disease label.
It is a forensic reconstruction of a biological pathway: identifying a failure in clearance, mapping the resulting substrate accumulation, tracing the systemic organ damage, and confirming the entire cascade through genomic and biochemical evidence.
When evaluating a suspected LSD case, look beyond single symptoms to the overarching pattern; look beyond single variants to the affected biochemical pathway; and look beyond genomic data to the total causality chain. When these connections are clearly forged, true clinical clarity is achieved.
Discover the Power of GEBRA Firsthand
If you are currently evaluating an unresolved case or a complex, suspected LSD profile, experience how GEBRA transforms your analysis.
Evaluate variant prioritization, real-time HPO phenotype matching, gene-disease evidence curation, and ACMG criteria classification within a singular, frictionless workflow. [Request a GEBRA Demo Today] and elevate your clinical genomics pipeline.
FAQ
Q1. What exactly are Lysosomal Storage Disorders (LSDs)?
LSDs are a group of rare inherited metabolic disorders characterized by defects in lysosomal enzymes or transport systems. This leads to the progressive accumulation of undegraded substrates within cells, causing cellular dysfunction that manifests as widespread, systemic damage across multiple organs, including the nervous system, heart, musculoskeletal system, liver, spleen, and kidneys.
Q2. Why is evaluating the “phenotype pattern” so critical in LSD cases?
LSDs frequently present with highly variable and non-textbook clinical manifestations. Patients with the same underlying genetic condition can exhibit entirely different organ involvement and rates of progression. Identifying a holistic pattern—such as combining hepatosplenomegaly with skeletal anomalies—allows interpreters to recognize a systemic storage disease process that an isolated symptom check would miss.
Q3. How do biomarkers influence the genomic interpretation of LSDs?
Biomarkers are key drivers of variant interpretation, rather than simple secondary data. Critical examples include Lyso-Gb3 (Fabry), Lyso-Gb1 (Gaucher), and urinary GAG (MPS). Reviewers should synthesize genetic variants, biomarkers, enzyme activity tests, and phenotype information into a unified causality chain.
Q4. Why is an NGS report alone often insufficient for a definitive LSD diagnosis?
While NGS is exceptionally powerful at identifying potential genetic variants, it cannot definitively confirm a metabolic diagnosis on its own, particularly in atypical or late-onset presentations. True diagnostic certainty requires validating that the genetic finding correlates precisely with phenotypic presentations, pathway mechanisms, and biochemical functional assays.
References
- Ferreira CR, Gahl WA. Lysosomal storage diseases. Transl Sci Rare Dis. 2017;2:1–71. doi:10.3233/TRD-160005.
- Hong X, Daiker J, Sadilek M, et al. Biomarker testing for lysosomal diseases: A technical standard of the ACMG. Genet Med. 2024;26(12):101245. PMID:39499245.
- Hall PL, Burlina A, Hahn S, et al. Measurement of lysosomal enzyme activities: A technical standard of the ACMG. Genet Med. 2022;24(7):1411–1418.
- Filocamo M, Morrone A. Biomarkers in Lysosomal Storage Diseases. Diseases. 2016;4(4):40. doi:10.3390/diseases4040040.
- Sanofi Genzyme. Gaucher Disease 101 — Lysosomal Storage Disorders.
- SSIEM ETAC 2024. Lecture 5 — Overview on Lysosomal Storage Disorders.
- Mapping Lysosomal Storage Disorders with Neurological Features by Cellular Pathways: Towards Precision Medicine. Curr Issues Mol Biol. 2025.
- Gelb MH, et al. Enzymatic screening and diagnosis of lysosomal storage diseases. CDC Stacks reference document.
- Burton BK, et al. Newborn screening for lysosomal storage diseases: current state, challenges, and implementation guidance.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the ACMG and AMP. Genet Med. 2015;17(5):405–424.
- Brain Sci. 2024;14(11):1085. doi:10.3390/brainsci14111085.
- Sanofi. Pompe Disease Patient Education Materials — Signs and Symptoms in IOPD/LOPD Patients.
- Sun A. Lysosomal storage disease overview. Ann Transl Med. 2018;6(24):476. doi:10.21037/atm.2018.11.39.
Get exclusive rare disease updates
from 3billion.

3billion Inc.
3billion is dedicated to creating a world where patients with rare diseases are not neglected in diagnosis and treatment.