How AI Prediction Tools Are Changing Rare Disease Diagnosis — ACMG PP3 Evidence Strength, REVEL, and 3Cnet
Quick Summary
- PP3 and BP4 are the primary ACMG/AMP criteria that rely on in silico tools for missense variant pathogenicity assessment.
- The 2022 ClinGen study (Pejaver et al.) demonstrated that PP3 can be applied at moderate or strong evidence strength — not just supporting — when score thresholds are met.
- Among the four recommended tools, REVEL is the only one capable of reaching both PP3 strong and BP4 very strong.
- At 3billion, REVEL is our primary tool, complemented by 3Cnet — our in-house predictor — to address the gaps REVEL cannot cover.
1. The Fastest-Moving Corner of Variant Interpretation
Interpreting a variant in rare genetic disease requires weighing evidence across a broad set of ACMG/AMP criteria — population frequency, variant type, segregation, functional studies, and more.
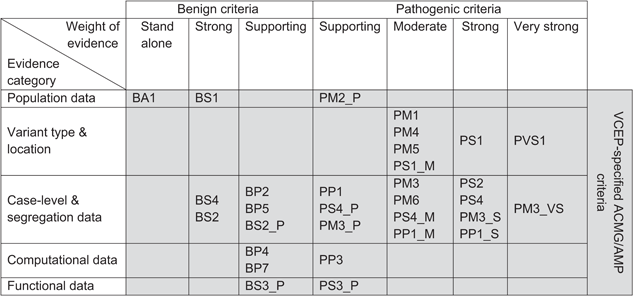
Within this framework, three criteria rely specifically on in silico computational tools: PP3, BP4, and BP7. Of these, PP3 and BP4 are grounded in missense variant pathogenicity prediction tools. This post focuses on PP3 and BP4.
- PP3: Applied when in silico tools predict that a variant will affect protein function (evidence toward pathogenicity)
- BP4: Applied when in silico tools predict that a variant will not affect protein function (evidence toward benignity)
The reason these two criteria are drawing increasing attention is straightforward: this is where advances in AI and deep learning are reflected most directly and most rapidly. Functional studies take time. Family history is not always available. But computational prediction models see new iterations every year — with improving accuracy. No other category of variant evidence is evolving at this pace.enicity prediction tools apply directly — and where AI advances are reflected most rapidly. Functional studies take time. Family history is not always available. But computational models improve every year, and their clinical weight is being actively redefined.
2. How It Used to Work — “Multiple Tools Agree = One Supporting Credit”
The 2015 ACMG/AMP guidelines defined PP3 and BP4 as follows:
“Multiple in silico tools supporting a deleterious or benign effect on the same variant can be recognized as supporting-level evidence.”
Under this framework, PP3 was fixed at a single supporting unit — regardless of how many tools were used or how high the scores were. Reaching LP required combining supporting criteria with stronger evidence, making PP3 alone a limited contributor to classification.
This approach had real problems:
- Many tools share overlapping training data, meaning their outputs are not truly independent lines of evidence
- Widely used tools like SIFT, PolyPhen-2, and CADD failed to meet the supporting threshold for PP3 even at developer-recommended scores
- CADD’s recommended threshold of 20.0 was found to fall within the BP4 moderate range — meaning it was being used as pathogenicity evidence incorrectly in many labs
3. The 2022 ClinGen Question — If Tools Differ in Reliability, Shouldn’t Evidence Strength Scale With the Score?
The ClinGen Sequence Variant Interpretation Working Group addressed this directly in a 2022 paper published in the American Journal of Human Genetics:
The team’s central question:
“When a tool’s score is sufficiently high, can PP3 be applied at moderate or strong strength — rather than always supporting?”
To test this, they built a carefully curated dataset from ClinVar (pathogenic/likely pathogenic and benign/likely benign variants; 11,834 variants, 1,914 genes), then applied a Bayesian probabilistic framework to identify score thresholds where each tool’s likelihood ratio met the criteria for supporting, moderate, strong, and very strong evidence.
The finding: evidence strength that each tool can reach varies.
Of the 13 tools evaluated, only 4 could reach PP3 strong. The remaining 9 topped out at moderate or supporting. GERP++ failed to meet even PP3 supporting.
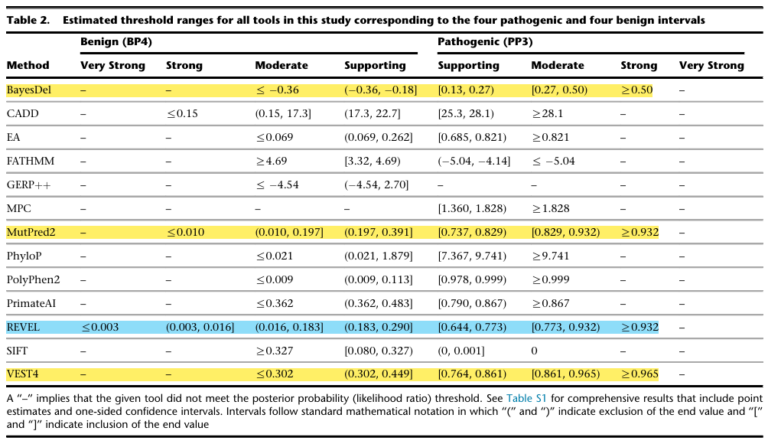
The paper’s recommendation to clinical laboratories:
“For most situations, clinical laboratories should use a single tool, genome-wide, that can reach the strong level of evidence for pathogenicity and moderate for benignity — BayesDel, MutPred2, REVEL, or VEST4 among the tools evaluated here.”
4. Why REVEL Became the Reference Standard
Among the four recommended tools, REVEL holds a uniquely strong position.
It is the only tool covering the widest evidence spectrum in both PP3 and BP4 directions. From PP3 strong (≥ 0.932) to BP4 very strong (≤ 0.003), no single tool matches its range.
One clarification is worth making explicit. The fact that REVEL can theoretically reach BP4 very strong is separate from whether that strength should be applied in practice. At 3billion, we scale PP3 evidence strength upward according to REVEL scores as recommended. For BP4, however, we apply only up to supporting — not moderate or strong. For genes that are not yet well-characterized, or for novel variants, a strong benign call risks filtering out a potential diagnosis before it can be properly evaluated. We prioritize preventing that risk over maintaining symmetry between the two directions. It is a deliberately conservative choice.
It is also worth noting that although REVEL is a meta-predictor integrating scores from multiple tools, the ClinGen study removed training variants from REVEL and all its constituent tools from the evaluation dataset — and its performance held. This addresses the most common concern about circularity in ensemble models.
And one more point: REVEL is a publicly distributed tool from NIH/NCBI. Being an openly available tool validated by an independent institution gives it a foundation of trust that matters in clinical settings.
5. What PP3 at Moderate or Strong Actually Means Clinically
The thresholds changed — but what does that actually mean for a diagnostic decision?
The ACMG/AMP classification system assigns points to each criterion and sums them to determine classification (Tavtigian et al., 2020):
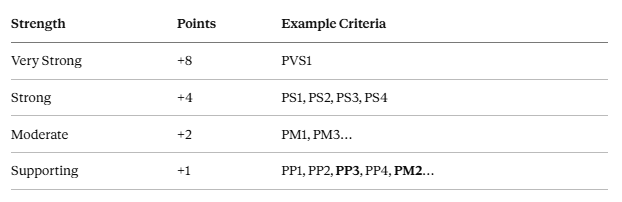
A note on PM2: The 2015 ACMG guidelines classified PM2 as moderate (+2). As gnomAD has expanded to hundreds of thousands of individuals, “absent from gnomAD” carries considerably less discriminatory weight than it once did. Many VCEPs now apply PM2 as PM2_Supporting (+1). 3billion follows this practice.
LP (Likely Pathogenic) threshold: ≥ +6 points / P (Pathogenic): ≥ +10 points
Under the original framework, PP3 was always supporting — a fixed +1 point. After the 2022 ClinGen update, PP3 scales with REVEL score:
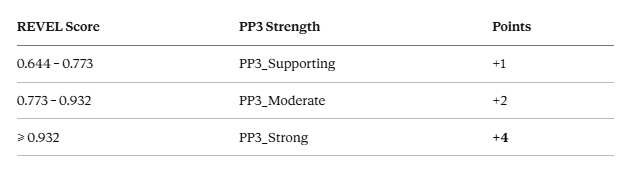
Here is what that difference means in practice.
[Scenario A] — Original 2015 framework (PP3 = always +1)
A missense variant identified in a patient with a rare genetic disease. Available evidence:

Two points short of LP. Despite a REVEL score of 0.95, the variant remains a VUS. PM2 (rarity) and PM1 (domain location) are both present, and REVEL is 0.95 — yet PP3 is capped at supporting (+1) and the variant goes nowhere.
[Scenario B] — After the 2022 ClinGen update (PP3_Strong, with PM1)
Same variant, same REVEL score of 0.95.

PP3 is now strong, but the variant is still VUS. Two reasons: First, when PP3_Strong is combined with PM1, their combined contribution cannot exceed strong (+4) (Pejaver et al., 2022, Discussion — Combining rules). PM1 already contributes +2, so PP3_Strong is capped at +2. Both criteria partially reflect evolutionary conservation, making this cap a necessary safeguard against double-counting. Second, with PM2 at supporting (+1), the total reaches only +5. That said, VUS-high (+5) sits one point from the LP threshold — a single additional line of evidence would tip the classification.
One more note: at 3billion, we extend this cap to PS1 and PM5 as well. PS1 (same amino acid change previously reported as pathogenic) and PM5 (different amino acid change at the same residue, previously pathogenic) are not feature-level overlaps with REVEL — they are clinical observations about specific positions. But combining PP3 with PS1 or PM5 effectively layers multiple pathogenicity signals on the same genomic location. We cap their combined contribution at strong (+4) as a conservative internal policy to prevent overweighting.
PP3_Strong therefore contributes its full +4 only when PM1, PS1, and PM5 are all absent.
[Scenario C] — PP3_Strong at full weight (no PM1/PS1/PM5, phenotype match)
A novel missense variant where PM1, PS1, and PM5 are all absent. The position-based overlap restriction does not apply, allowing PP3_Strong its full +4.

What tips the classification here is not REVEL — it is phenotype (PP4). Even with PP3_Strong at full +4, PM2_Supporting (+1) brings the total to only +5 — VUS-high. PP4 applies when the patient’s clinical presentation is sufficiently specific to the gene-disease association, and that final point crosses the LP threshold. Without phenotype, the same variant stays VUS-high, which should be read as a signal that functional evidence or family segregation data is needed. Whether PP4 can be applied is a clinical judgment that belongs to the treating geneticist.
6. REVEL’s Limitations
REVEL is powerful, but has two important limitations.
First — Scope
REVEL applies only to missense variants. Splice site variants, non-coding regulatory variants, frameshifts, and stop-gain variants receive no REVEL score. Beyond variant type, every tool including REVEL falls into an indeterminate zone for 26–50% of variants (Pejaver et al., 2022, Figure 4B). A substantial proportion of variants simply cannot be assessed by REVEL.
Second — Structural Bias Toward ClinVar
REVEL is an ensemble model, and the tools that comprise it are trained predominantly on ClinVar data. Although the ClinGen study removed training variants from the evaluation set, the underlying models reflect patterns repeatedly seen in ClinVar — specific genes, common disease associations, recurring variant types. REVEL performs strongly where ClinVar data is rich, but may be less reliable for data-sparse genes or novel variant patterns not yet captured in public databases.
This is partly why models like AlphaMissense and EVE — trained on protein sequence and evolutionary information without relying on ClinVar labels — are attracting attention. Because they do not depend on what has already been classified, they offer a different kind of signal, potentially better suited to identifying genuinely novel pathogenic variants.
7. 3Cnet — 3billion’s Complementary Tool
To address these gaps, 3billion developed 3Cnet.
3Cnet is not an ensemble of existing tools. It is a Recurrent Neural Network (RNN)-based deep learning model that learns directly from amino acid sequence context. Key design features:
- Multi-task learning: Trained simultaneously on ClinVar clinical data, gnomAD common variants (as benign proxies), and evolutionary conservation data derived from multiple sequence alignments (MSA) of the UniRef database — reducing overfitting to any single data source
- No circularity: 3Cnet uses no scores from other prediction tools as input, making it structurally independent from REVEL
- Broader variant scope: Applicable to non-missense variants including start-lost, stop-gain, frameshift, and deletion variants
Performance (external validation, ClinVar August 2020):
On an independent dataset of 6,298 pathogenic and 6,468 benign variants, 3Cnet achieved ROC-AUC of 0.925, compared to REVEL’s 0.910 — the highest among all tools evaluated. Subsequent versions have improved further: 3Cnet v2.3 reached 0.946, and v2.5 reached 0.975.
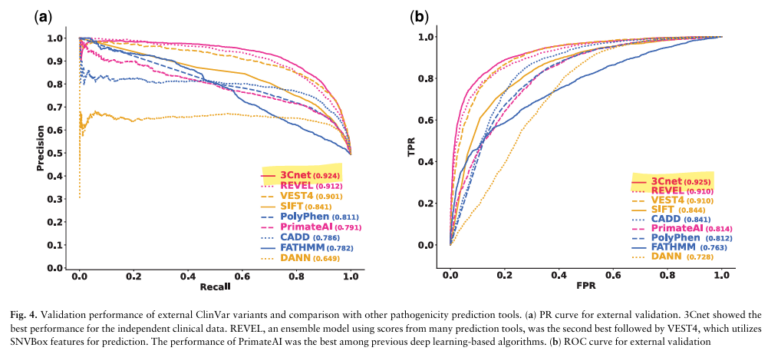
A more clinically meaningful metric is top-k recall — the probability of ranking the true disease-causing variant within the top k candidates in a real patient genome. In confirmed rare disease patients (n=111), 3Cnet’s top-1 recall was 2.2× higher than REVEL’s. A patient genome typically contains 100–400 rare missense variants; identifying the true causal variant as the top-ranked candidate is roughly twice as likely with 3Cnet.
A note on caution in clinical application
These performance numbers are encouraging — but they are also why 3Cnet is positioned as a complementary tool, not a primary classifier.
The 3Cnet authors are explicit about this in the paper itself:gs to the clinical geneticist.
“3Cnet was trained on benign variants mainly from public databases such as ClinVar and gnomAD, a composition that is not free from bias. 3Cnet could misunderstand some benign variants as pathogenic if such variants were not reported in the databases. Therefore, using 3Cnet to identify benign human variants needs to be careful and rigorous validation should be followed.” (3Cnet paper, Discussion)
REVEL, on the other hand, is one of the four tools formally recommended by ClinGen, with an extensively validated evidence base across the clinical community. From the standpoint of diagnostic consistency and reproducibility, maintaining REVEL as the primary standard is the rational choice.
8. 3billion’s Approach — REVEL First, 3Cnet as a Second Layer
In practice, 3billion’s variant interpretation workflow works as follows.
REVEL is applied first, using ClinGen-recommended score thresholds to assign PP3/BP4 evidence strength. This maintains consistency with clinical standards.
3Cnet is applied when REVEL cannot provide sufficient guidance — when REVEL scores fall in the indeterminate range, or for variant types outside REVEL’s scope.
The real-world impact of this approach is illustrated in 3Cnet’s Table 1. Excluding variants already covered by REVEL (score ≥ 0.75), the team identified 7 novel variants with 3Cnet scores ≥ 0.9 where REVEL could not provide strong evidence. Among these, 1 variant (NP_056651.1: p.Gly54Glu) could only be reclassified from VUS to Likely Pathogenic when 3Cnet’s score was applied as PP3 evidence.
One case. But in rare disease diagnosis, the difference between “VUS” and “Likely Pathogenic” for a single patient changes the trajectory of their care — and the testing decisions for their entire family.
Expert Perspectives
To ground these concepts in clinical practice, we spoke with two members of 3billion’s team.
Clinical Team Lead Interview Chief Medical Officer / Gohun Seo, MD, PhD
Q: Has the updated PP3 framework changed how you approach variant classification in practice?
“Before 2022, a REVEL score of 0.95 still only gave you one supporting credit. If other evidence was thin, the variant stayed a VUS regardless of the score. After ClinGen 2022, PP3 can now register as moderate or even strong — that’s a real change. But that doesn’t mean LP automatically follows. Because we apply PM2 as supporting, even PP3_Strong usually lands us at VUS-high, and whether we go further depends entirely on what other evidence is there. Variants that already have strong evidence like PS1 get reported even when the referral doesn’t come with detailed phenotype documentation — and honestly, that’s the more common scenario. On the flip side, when all you have is a computational score and rarity, clinical phenotype becomes decisive. The stronger the tools get, the more important it is to have someone who knows how to weigh each piece of evidence.”
Q: How do you use 3Cnet alongside REVEL in real cases?
“REVEL comes first, and 3Cnet is second. We look at 3Cnet when REVEL is indeterminate or the score is ambiguous. When both tools agree, the evidence feels more solid. When they diverge, that’s a signal to slow down and look more carefully. Having two independent perspectives is a safety net — better than relying on one tool alone.”
3Cnet Development Lead Interview Chief Scientific Officer / Kyoungyeul Lee, PhD
Q: What was the central challenge you focused on when building 3Cnet?
“Circularity. Meta-predictors like REVEL use scores from other tools as inputs, which means many tools are effectively looking at the same information repackaged. 3Cnet doesn’t use any external tool scores at all — it learns entirely from amino acid sequences and evolutionary conservation information. That’s what allows REVEL and 3Cnet to have genuinely independent perspectives on the same variant.”
Q: The performance numbers are impressive — where does caution still apply in clinical use?
“We wrote it in the paper directly: 3Cnet is not free from bias. Because it was trained on ClinVar and gnomAD, variant types or specific ethnic populations that aren’t well-represented in those databases may see different performance. That’s part of why we position 3Cnet as a supplementary tool. A high 3Cnet score is a signal to look more carefully — not a basis for confirming a diagnosis on its own. That judgment belongs to the clinical geneticist. Our role is to provide good information so that judgment can be made as well as possible.”
Closing
PP3 and BP4 sit at the intersection of clinical variant interpretation and rapidly advancing AI — and the 2022 ClinGen update made that intersection more consequential. What was once a fixed supporting credit can now, under the right conditions, carry the weight of a strong criterion.
But as this post has shown, the same PP3_Strong score plays out very differently depending on what else is present. REVEL at 0.95 does not automatically produce an LP classification — whether PM1 is there, whether PM2 is applied as supporting, whether the patient’s phenotype provides corroborating evidence all matter. Stronger tools raise the ceiling of what’s possible; they do not remove the need for expert judgment about how to use what they provide.
This is what distinguishes clinical genetic diagnosis from algorithmic scoring. Generating a number and interpreting what that number means in a specific clinical context are fundamentally different tasks. 3billion develops tools like 3Cnet and integrates REVEL with one goal: to give clinical geneticists and genetic counselors better raw material for the judgments that only they can make. That is the direction in which we continue to build.
For questions about rare disease diagnosis or variant interpretation, please reach out.
References
- Pejaver V, et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet. 2022;109(12):2163–2177. https://doi.org/10.1016/j.ajhg.2022.10.013
- Won D-G, Kim D-W, Woo J, Lee K. 3Cnet: pathogenicity prediction of human variants using multitask learning with evolutionary constraints. Bioinformatics. 2021;37(24):4626–4634. https://doi.org/10.1093/bioinformatics/btab529
- Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424.
https://pubmed.ncbi.nlm.nih.gov/25741868/ - Tavtigian SV, et al. Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. Hum Mutat. 2020;41(10):1734–1737. https://doi.org/10.1002/humu.24088
Get exclusive rare disease updates
from 3billion.

Sookjin Lee
Expert in integrating cutting-edge genomic healthcare technologies with market needs. With 15+ years of experience, driving impactful changes in global healthcare.





