Episode 6 – Cornelia de Lange syndrome (CdLS): Beyond the Variant
📌 Series Introduction
이 시리즈는 3billion의 임상 유전학 전문가가 직접 작성한 글입니다. AI 기반 유전변이 판독 도구가 보편화된 시대에, 자동화된 우선순위화 이후 판독자가 실제로 무엇을 이해하고 어떻게 판단해야 하는지에 초점을 맞춥니다.
각 편에서는 하나의 질환군을 중심으로, 유전적 기전과 임상 스펙트럼을 함께 살펴보며 변이를 “찾는 것”을 넘어 “설명하고 해석하는” 데 필요한 관점을 공유하고자 합니다.
Cornelia de Lange syndrome (CdLS) 정리
Key Takeaway
CdLS는 phenotype으로 의심하고, variant 해석으로 확정되는 질환이며, 그 해석이 축적될 때 비로소 반복 가능한 진단이 됩니다.
CdLS와 같은 질환에서는 단순히 variant를 찾는 것보다 “해석을 어떻게 구조화하고 남기느냐”가 더 중요합니다.
CdLS란 무엇인가?
Cornelia de Lange syndrome (CdLS)는 오래전부터 알려진 희귀 유전 질환입니다.
1849년에 처음 보고되었고, 1930년대 Cornelia de Lange에 의해 임상적으로 정리되었습니다. 이후 2004년에 NIPBL 유전자가 발견되면서 분자적 원인이 밝혀졌습니다.
👉 즉, 임상적으로 먼저 정의되고 이후 유전적 원인이 밝혀진 질환입니다.
임상에서는 어떻게 보일까요?
CdLS는 여러 시스템에 걸쳐 나타나는 전형적인 syndromic disorder입니다.
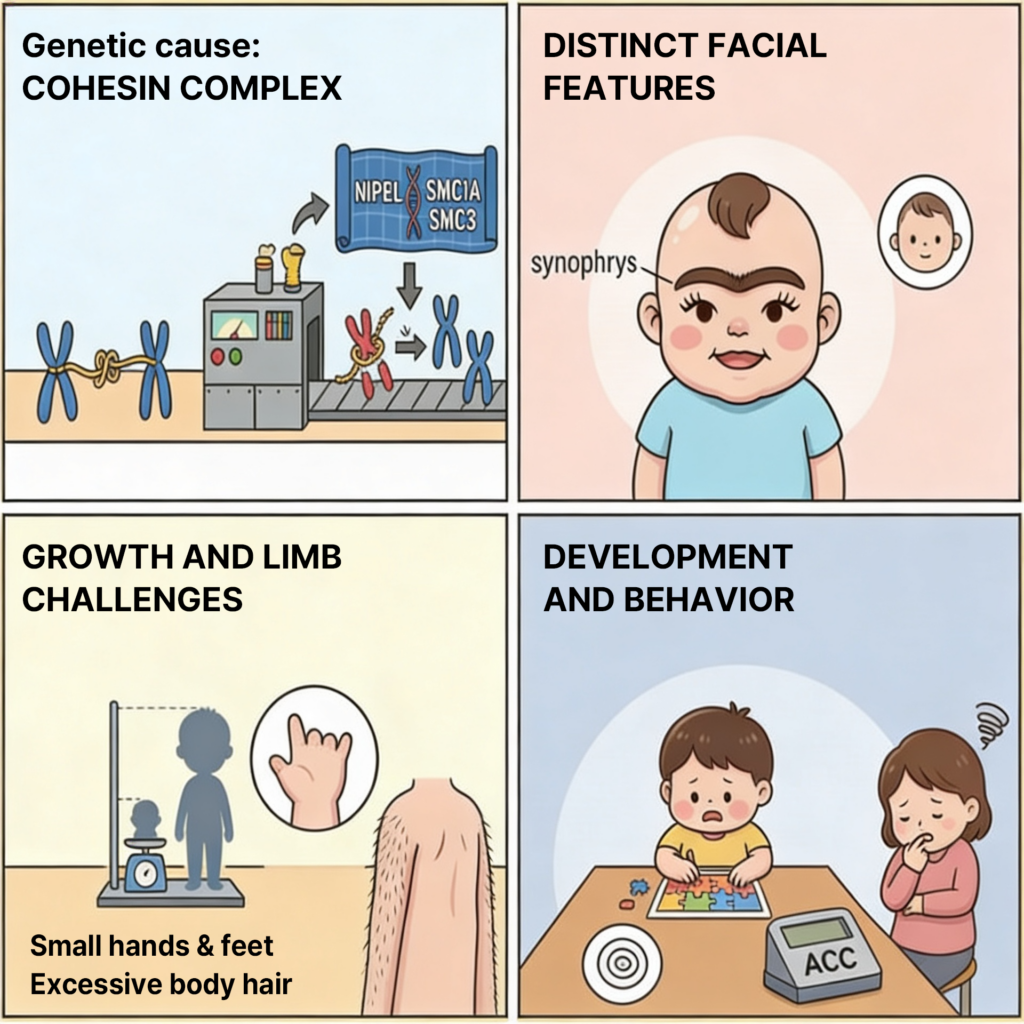
대표적인 얼굴 특징으로는 synophrys, hirsutism, anteverted nares가 있으며 이는 CdLS를 의심하게 만드는 중요한 단서입니다.
여기에 성장 지연과 신경발달 이상, 그리고 self-injurious behavior가 함께 나타나는 경우가 많습니다.
또한 upper limb 이상, GERD, 선천성 심장 질환, 청력 이상 등 다양한 장기 이상이 동반될 수 있습니다.
👉 정리하면 CdLs의 임상적 특징은 얼굴 + 성장/발달 + 사지 이상 이 세 가지 축으로 이해하시면 좋습니다.
그런데 이것만으로 진단이 가능할까요?
과거에는 특정 얼굴/임상 특징 중심으로 진단이 되었습니다.
이런 특징이 확인되면 “이 정도면 CdLS 아닐까?”라고 생각했습니다.
하지만 실제 임상에서는 아래와 같은 특징때문에 진단으로 연결되지 못하는 경우가 있었습니다.
- 표현형이 매우 다양하고
- 전형적이지 않은 케이스도 많으며
- 다른 질환처럼 보이는 경우도 많습니다
유전자 연구가 진행되면서 CdLS는
❗ 눈으로 보고 확정하는 질환이 아니라
❗ 유전 변이까지 확인해야 진단이 완성되는 질환입니다
왜 이렇게 다양하게 보일까요?
현재 CdLS는 cohesinopathy, 즉 스펙트럼 질환으로 이해됩니다.
- 같은 유전자에서도 다양한 phenotype
- 다른 유전자에서도 유사한 phenotype 이 나타날 수 있습니다.
그래서 Classic / Non-classic / CdLS-like라는 개념이 등장했습니다.
질환의 본질은 “전사 조절 이상”입니다
CdLS의 핵심은 cohesin 자체가 아니라 transcription regulation입니다.
- chromatin 구조 변화
- enhancer–promoter interaction 붕괴
- 전반적인 유전자 발현 변화
👉 결과적으로 전체 발현 패턴이 미세하게 흐트러지는 상태가 됩니다
이 때문에 phenotype이 일정하지 않게 나타납니다.
유전자별로 보면 조금 더 명확해집니다
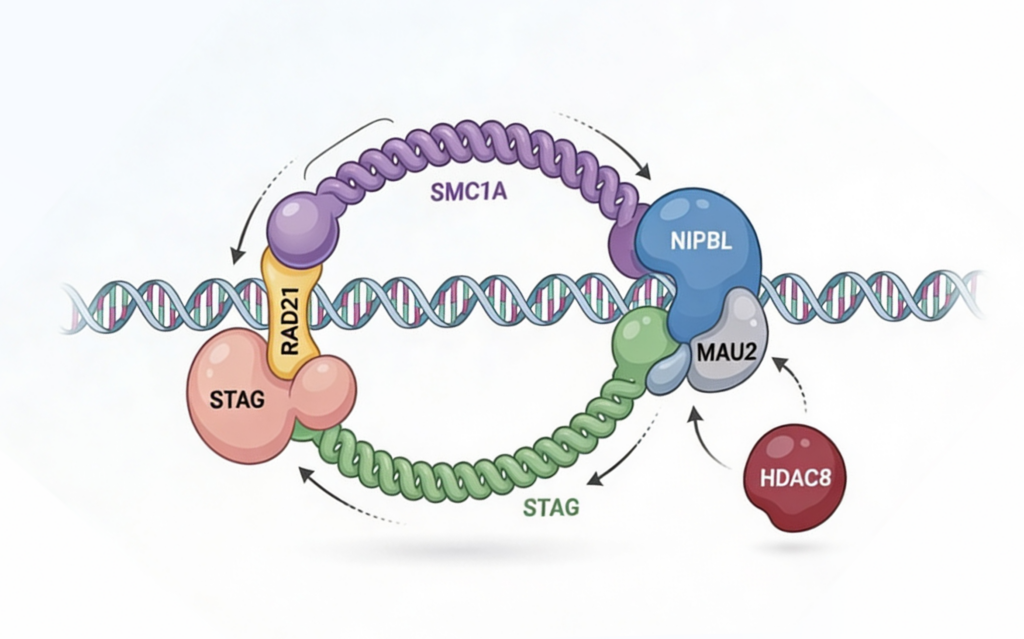
CdLS는 여러 유전자와 연관되어 있으며, 각 유전자에 따라 어느 정도 phenotype 경향이 나타납니다.
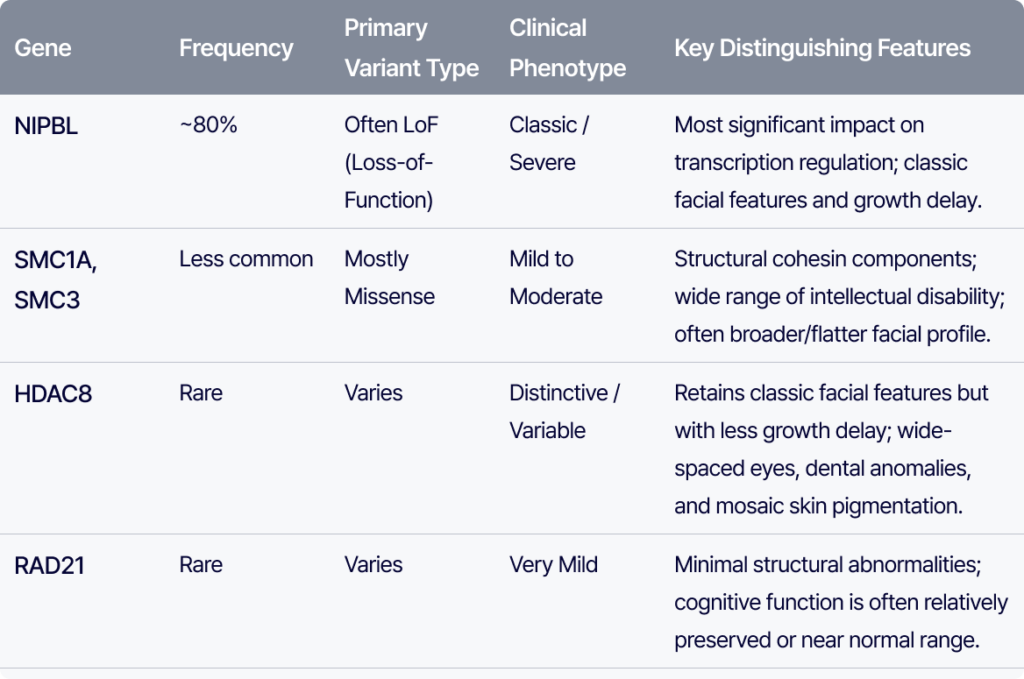
가장 흔한 원인은 NIPBL (~80%)입니다. 주로 LoF 변이가 많으며, 전사 조절에 큰 영향을 주기 때문에 👉 비교적 classic하고 severe한 phenotype을 보이는 경우가 많습니다.
반면 SMC1A, SMC3는 cohesin 구조를 유지하는 단백질로, 대체로 missense, in-frame deletions 변이가 많고 지적 장애(ID)는 관찰되며, 중등도에서 중증까지 다양하게 나타납니다. 또한 눈썹은 약간 더 평평하고 넓은 형태를 보이며, 콧등은 더 넓고 길게 나타납니다.
HDAC8의 경우 CdLS facial feature는 유지되지만 성장 지연은 상대적으로 심하지 않은 경우가 많습니다. 넓은 눈 간격, 넓은 코, dental anomaly, 그리고 mosaic skin pigmentation 같은 특징이 힌트가 됩니다.
RAD21은 구조적 이상이 거의 없고 인지 기능도 비교적 보존되는 경우가 많습니다. 일부는 정상 범위에 가까운 경우도 보고됩니다.
👉 정리하면
- NIPBL → severe 경향
- SMC 계열 → mild–moderate
- HDAC8 → moderate
- RAD21 → 매우 mild
하지만 이 구분은 절대적인 기준이 아니라 “경향”일 뿐이라는 점이 중요합니다.
그래서 판독이 어려워집니다
실제 임상에서는 다음과 같은 상황이 자주 발생합니다.
- 관련 유전자의 variant는 확인
- phenotype이 애매
- 확신이 부족한 상태
또는 ‘같은 데이터를 두고도 팀마다 결론이 달라지는 경우’
특히 CdLS처럼 “설명이 필요한 질환”에서 이 문제가 더 크게 나타납니다.
결국 남는 질문 하나
CdLS에서 가장 중요한 질문은 이것입니다.
👉 “이 변이가 왜 이 환자를 설명하는가?”
이 질문에 답하려면 해석 자체가 구조화되어야 합니다
- phenotype–gene 연결
- functional impact 해석
- differential과의 비교
여기서 많은 팀이 막힙니다
문제는 보통 이 단계에서 발생합니다.
- 해석은 했지만 기록이 남지 않고
- 같은 variant를 매번 새롭게 해석하며
- 팀 내에서도 판단 기준이 달라집니다
👉 결과적으로 해석은 개인의 경험에 의존하게 됩니다.
판독자 Tip
- phenotype은 출발점일 뿐입니다
- 전형적이지 않은 케이스를 쉽게 배제하지 마십시오
- CdLS-like 질환을 항상 함께 고려하십시오
👉 그리고 가장 중요한 것: 해석은 반드시 기록되어야 합니다.
한 줄 정리
👉 CdLS는 phenotype으로 의심하고, variant 해석으로 확정되는 질환이며, 그 해석이 축적될 때 비로소 반복 가능한 진단이 됩니다.
이 글에서 다룬 것처럼, CdLS와 같은 질환에서는 단순히 variant를 찾는 것보다 “해석을 어떻게 구조화하고 남기느냐”가 더 중요합니다.
💡 혹시 “이런 케이스들을 좀 더 구조적으로 풀 수는 없을까?”라는 생각이 들었다면?
GEBRA는
- phenotype–variant 연결
- 해석 근거 기록
- 반복 가능한 interpretation workflow
를 통해 판독 과정을 더 빠르고 일관되게 만드는 것을 목표로 합니다.
References
- Parenti I, Kaiser FJ. Cornelia de Lange syndrome as paradigm of chromatinopathies. Front Neurosci. 2021;15:774950. doi:10.3389/fnins.2021.774950
- Deardorff MA, Noon SE, Krantz ID. Cornelia de Lange syndrome. In: Adam MP, Bick S, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2026 [updated 2020 Oct 15]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1104/
Sarogni P, Pallotta MM, Musio A. Cornelia de Lange syndrome: from molecular diagnosis to therapeutic approach. J Med Genet. 2020;57(5):289–295. doi:10.1136/jmedgenet-2019-106277 - Minor A, Shinawi M, Hogue JS, Vineyard M, Hamlin DR, Tan C, Donato K, Wysinger L, Botes S, Das S, Del Gaudio D. Two novel RAD21 mutations in patients with mild Cornelia de Lange syndrome-like presentation and report of the first familial case. Gene. 2014;537(2):279–284. doi:10.1016/j.gene.2013.12.045
- Santen GW, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M, Kant SG, Snoeck IN, Peeters EA, Hilhorst-Hofstee Y, Wessels MW, den Hollander NS, Ruivenkamp CA, van Ommen GJ, Breuning MH, den Dunnen JT, van Haeringen A, Kriek M. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet. 2012;44(4):379–380. doi:10.1038/ng.2217
- Raible SE, Mehta D, Bettale C, Fiordaliso S, Kaur M, Medne L, Rio M, Haan E, White SM, Cusmano-Ozog K, Nishi E, Guo Y, Wu H, Shi X, Zhao Q, Zhang X, Lei Q, Lu A, He X, Okamoto N, Miyake N, Piccione J, Allen J, Matsumoto N, Pipan M, Krantz ID, Izumi K. Clinical and molecular spectrum of CHOPS syndrome. Am J Med Genet A. 2019;179(7):1126–1138. doi:10.1002/ajmg.a.61174
3billion 뉴스레터 구독자만을 위한
희귀질환 진단 최신 정보를 받아보세요.

3billion Inc.
희귀질환 환자들이 진단과 치료에서 외면받지 않는 세상을 만들기 위해 쓰리빌리언은 하나의 미션을 바라봅니다.





