Marfan Syndrome: A Summary of Genetic Causes and Ghent Criteria
📍Key Takeaways
- Genetics: Marfan syndrome is caused by FBN1 pathogenic variants in ~90% of cases; TGFBR1/2 and FBN2 variants account for overlapping phenotypes.
- Diagnosis: Follows the Revised Ghent Nosology (2010, the global diagnostic standard for Marfan syndrome), weighted heavily toward aortic root dilation and ectopia lentis.
- Mortality: Aortic dissection (primarily Type A) is the leading cause of mortality — regular echocardiographic surveillance is mandatory.
- Genetic Testing: Indicated when clinical criteria are equivocal, for family cascade screening, and to guide reproductive decisions.
- Prognosis: Genotype–phenotype correlations carry meaningful implications for aortic risk stratification.

Marfan syndrome is a systemic connective tissue disorder caused by pathogenic variants in the FBN1 gene. Affecting approximately 1 in 5,000 individuals worldwide, it carries life-altering cardiovascular risks that are largely preventable with timely diagnosis and surveillance. Yet diagnostic delay — averaging 5 to 7 years in published cohorts — remains a persistent clinical challenge.
This review is written for clinicians who encounter suspected Marfan syndrome in practice and need a concise, evidence-based framework for diagnosis, genetic evaluation, and management.
1. What Is Marfan Syndrome?
Marfan syndrome (MFS) is an autosomal dominant disorder of connective tissue involving the cardiovascular, skeletal, and ocular systems. It was first described by Antoine Marfan in 1896, but its molecular basis — pathogenic variants in FBN1 encoding fibrillin-1 — was established by Dietz and colleagues in 1991.
The global prevalence is estimated at 1 in 5,000–10,000, with no sex or ethnic predilection. Approximately 75% of cases are inherited from an affected parent; the remaining 25% arise from de novo variants. While penetrance is complete, clinical expressivity is highly variable—even among affected family members.

2. Genetic Basis: FBN1 and Beyond
The FBN1 gene on chromosome 15q21.1 encodes fibrillin-1, a glycoprotein critical to the structural integrity of the extracellular matrix (ECM). Over 3,000 pathogenic variants have been described, with missense mutations (where a single nucleotide change results in a different amino acid) being the most common.
Fibrillin-1 dysfunction results in:
- Structural weakness of elastic tissue (aorta, lens zonules, periosteum).
- Dysregulation of TGF-β signaling, driving aortic wall remodeling.
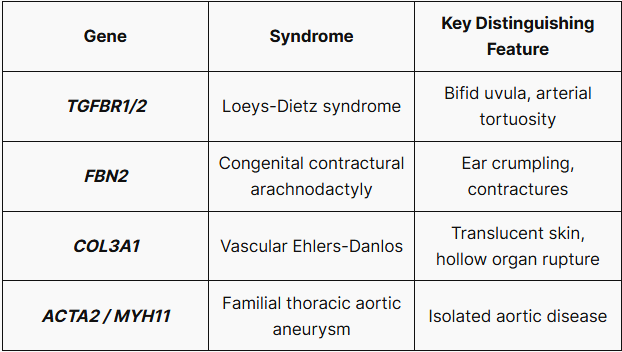
Differential Diagnosis (Genetic Overlap)
3. Clinical Features by System
Cardiovascular (Life-threatening)
- Aortic root dilation / aneurysm (present in ~80% of adults)
- Aortic dissection (Type A at sinuses of Valsalva)
- Mitral valve prolapse (~75%), with risk of regurgitation
- Pulmonary artery dilation
Skeletal
- Tall stature with disproportionate limb length (dolichostenomelia)
- Arachnodactyly: Positive Steinberg thumb sign, Walker-Murdoch wrist sign
- Pectus excavatum or carinatum
- Scoliosis (>40° in severe cases)
Ocular
- Ectopia lentis (lens subluxation): Present in ~60%; superotemporal displacement is characteristic
- Myopia (often severe, >3 diopters)
- Increased risk of retinal detachment
Other
- Spontaneous pneumothorax (~10%)
- Dural ectasia (lumbosacral): Present in >60%; may cause chronic low back pain
4. Diagnostic Criteria: Revised Ghent Nosology (2010)
The 2010 revised Ghent Nosology places greater weight on aortic root dilation and ectopia lentis.
Systemic Score (Score ≥7 is significant)
| Feature | Points |
| Wrist AND thumb sign | 3 |
| Wrist OR thumb sign (one positive only) | 1 |
| Pectus carinatum | 2 |
| Pectus excavatum or chest asymmetry | 1 |
| Hindfoot deformity | 2 |
| Flat pes planus | 1 |
| Spontaneous pneumothorax | 2 |
| Dural ectasia | 2 |
| Protrusio acetabuli | 2 |
| Reduced US/LS ratio AND arm span/height | 1 |
| Scoliosis or thoracolumbar kyphosis | 1 |
| Reduced elbow extension | 1 |
| Facial features (3 of 5) | 1 |
| Skin striae | 1 |
| Myopia >3D | 1 |
| Mitral valve prolapse (MVP) | 1 |
Diagnosis is established when (according to Ghent Nosology):
- Aortic root dilation (Z-score ≥2) + ectopia lentis
- Aortic root dilation + FBN1 pathogenic variant
- Aortic root dilation + systemic score ≥7
- Ectopia lentis + FBN1 variant previously associated with aortic dilation
5. When and Why to Order Genetic Testing
Genetic testing is strongly indicated in the following scenarios:
- Equivocal presentation: Systemic score is on the borderline or lacks cardinal clinical features.
- Pediatric patients: Early diagnosis before clinical features fully manifest.
- Family cascade screening: Efficient testing for first-degree relatives.
- Atypical phenotype: To distinguish MFS from Loeys-Dietz or Vascular EDS.
- Reproductive planning: PGT or prenatal diagnosis.
6. Management and Surveillance
- Cardiovascular Monitoring: Perform echocardiography annually if the aortic root diameter is <4.5 cm, and every 6 months if it is ≥4.5 cm or showing rapid expansion.
- Lifestyle & Activity: Avoid competitive or contact sports and isometric exercises (e.g., heavy lifting, weightlifting) to prevent sudden blood pressure spikes.
- Pregnancy & Reproductive Care: Pregnancy is considered a relative contraindication if the aortic root exceeds 4.0 cm due to high risk. (Multidisciplinary management is mandatory for pregnancy planning and care.)
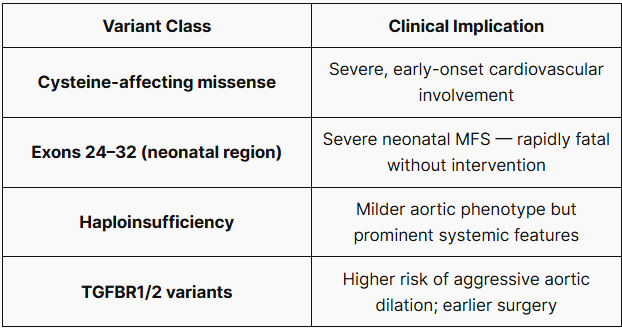
7. Genotype–Phenotype Correlation
8. Conclusion
Marfan syndrome is a preventable cause of premature aortic dissection. The cornerstone of care is early, accurate diagnosis using the Revised Ghent Nosology, followed by lifelong aortic surveillance and pharmacologic risk reduction. Genetic testing is no longer just a luxury — it is a standard tool for diagnosis, prognosis, and family planning.
For complex differential diagnosis, 3billion’s genetic testing services with automated reanalysis can help you find the answers. Contact us to learn more about our WES and WGS testing.
References
- Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476-485. https://doi.org/10.1136/jmg.2009.072785
- Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337-339. https://doi.org/10.1038/352337a0
- Judge DP, Dietz HC. Marfan’s syndrome. Lancet. 2005;366(9501):1965-1976. https://doi.org/10.1016/S0140-6736(05)67789-6
- Pyeritz RE. Marfan syndrome: improved clinical history results in expanded natural history. Genet Med. 2019;21(8):1683-1690. https://doi.org/10.1038/s41436-018-0399-4
- Faivre L, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations. Am J Hum Genet. 2007;81(3):454-66. https://doi.org/10.1086/520125
- Isselbacher EM, Ouriel K, Silberbach M, et al. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2022;80(24):e223-e393. https://doi.org/10.1016/j.jacc.2022.08.004
Get exclusive rare disease updates
from 3billion.

Soo-jung Baek
As a marketer, I strive to empower the rare disease community by sharing meaningful insights backed by our company’s expertise.