Episode 7: Prader-Willi and Angelman Syndromes — Beyond the Variant
📌 Series Introduction
This series is authored by clinical genetics experts at 3billion. In an era where AI-driven variant prioritization has become commonplace, we focus on what interpreters truly need to understand and how they should exercise clinical judgment after automated tools have done their part.
Each episode centers on a specific disease group, exploring genetic mechanisms alongside the clinical spectrum. Our goal is to share perspectives that go beyond simply “finding” a variant to “explaining and interpreting” it.
Key Takeaway
- Both Prader-Willi Syndrome (PWS) and Angelman Syndrome (AS) are disorders associated with the 15q11-q13 imprinting region.
- The fundamental difference between the two lies in the parent-of-origin effect rather than the genomic location itself.
- PWS is primarily associated with the loss of paternal gene expression, with SNORD116 playing a critical role.
- AS is driven by the loss of maternal UBE3A function within neurons.
- Variant interpretation for imprinting disorders must integrate information from the phenotype, methylation status, and inheritance patterns.
Same 15q11-q13, Different Conclusions: Lessons from PWS and Angelman Syndrome
Prader-Willi Syndrome (PWS) and Angelman Syndrome (AS) are representative imprinting disorders involving the 15q11-q13 region. Interestingly, while both conditions originate from the same chromosomal area, their clinical presentations are entirely distinct.
The key factor creating this difference is not merely “which region was lost,” but which parental allele was affected. Loss of function in the paternally derived 15q11-q13 region leads to PWS, whereas loss of function in the maternally derived UBE3A gene results in AS.
Ultimately, these two disorders pose a critical question to the interpreter: Is it enough to ask, “Where is this variant or CNV located?” Or must we also ask, “In what genetic and epigenetic context does this change lead to disease?”
Why do PWS and AS manifest so differently?
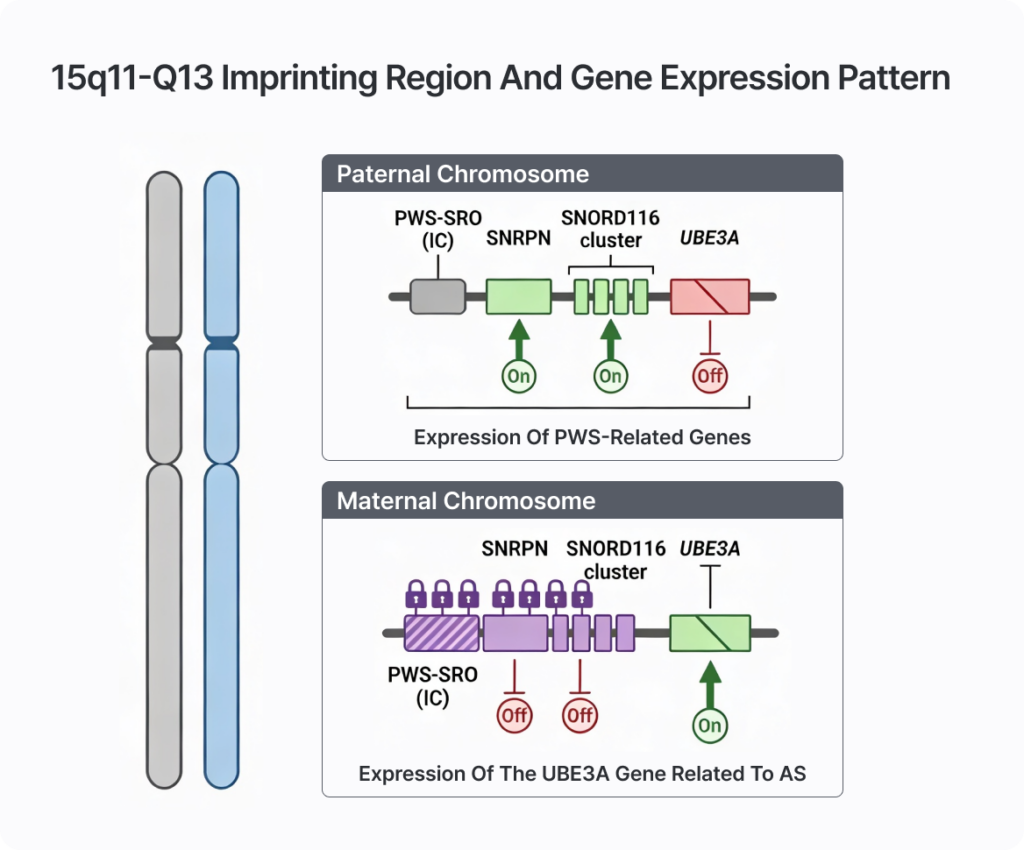
Differences in Imprinting Regulation and Transcription Initiation
The 15q11-q13 region is a classic imprinting region where different genes are expressed depending on their parental origin. Even with identical DNA sequences, gene expression depends on whether the chromosome was inherited from the father or the mother. This parent-of-origin specific expression is regulated by epigenetic mechanisms, primarily DNA methylation.
The “Imprinting Center” (IC), known as the PWS-SRO (Prader-Willi Syndrome Smallest Region of Overlap), plays a pivotal role here. This area is the critical minimal region of deletion for PWS and serves as the essential regulatory segment for the initiation of SNRPN transcription.
According to current models, a major reason for the differing imprinting patterns is the difference in the transcription start site. On the paternal chromosome, transcription begins at the standard exon; however, on the maternal chromosome, it starts further upstream at “alternative exon 1.” During this process, CpG islands within the PWS-SRO play a vital role.
Specifically, transcription initiated from alternative exon 1 on the maternal side acts as a signal that induces methylation in the adjacent PWS-SRO. While the exact mechanism is not yet fully elucidated, the prevailing model suggests that the transcription process itself alters chromatin structure, creating an environment conducive to methylation.
In other words, as RNA polymerase passes through this region, it modifies chromatin accessibility and nucleosome organization. This allows DNA methyltransferase to access the area more easily, inducing CpG island methylation. Indeed, reports show that removing the alternative exon 1 on the maternal side prevents the normal establishment of imprinting methylation. Thus, a very small difference in the transcription start site ultimately determines the methylation state and the overall gene expression pattern.
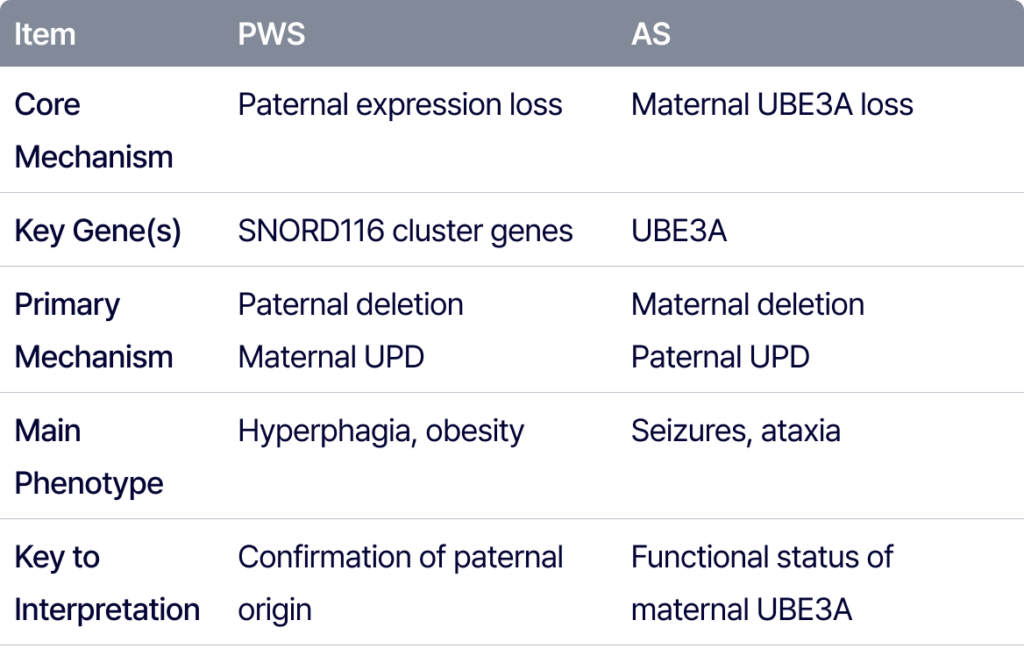
Paternal Expression Loss and the Pathophysiology of PWS
On the paternal chromosome, the PWS region promoter remains unmethylated, allowing for the active expression of several paternally expressed genes, including SNRPN and the SNORD116 cluster. Conversely, on the maternal chromosome, this promoter region is kept in a methylated state, suppressing the transcription of these genes.
SNORD116, in particular, is a core region that plays a significant role in the hyperphagia and hypothalamic dysfunction characteristic of PWS. Research on Snord116 deletion mouse models has shown a replication of the overeating and metabolic abnormalities seen in PWS patients.
UBE3A and the ‘Collision Model’ of Angelman Syndrome
In Angelman Syndrome (AS), the UBE3A gene takes center stage. While UBE3A is biallelically expressed in most tissues, it is functionally expressed primarily from the maternal allele in the nervous system, specifically in neurons.
This occurs because a long UBE3A-ATS (antisense transcript) is transcribed on the paternal chromosome. This antisense transcript is transcribed in the opposite direction of UBE3A, effectively suppressing paternal UBE3A expression.
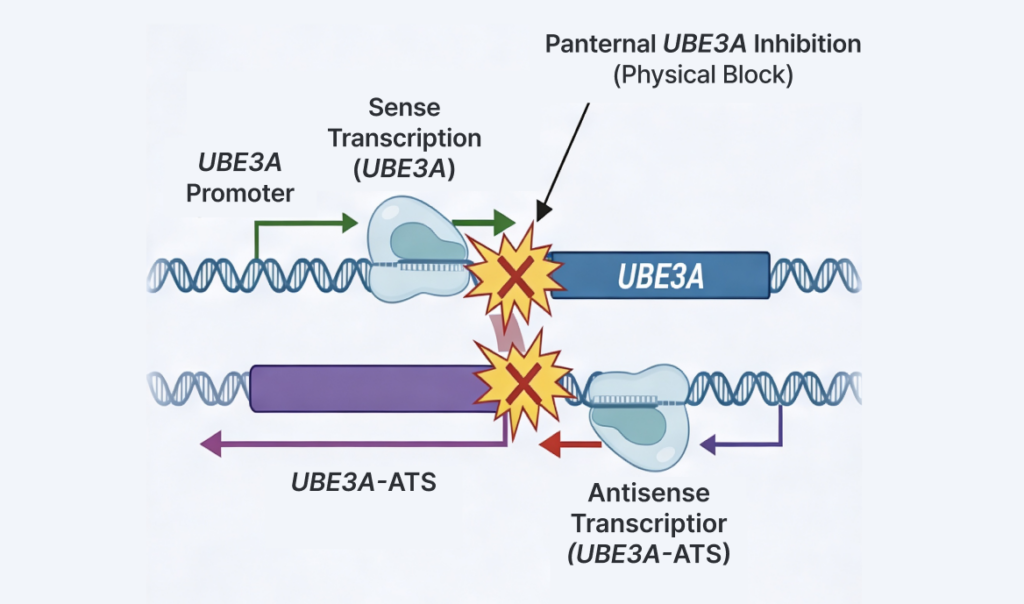
One of the best-known explanatory models for this is the “collision model.” In this model, the sense transcription of UBE3A and the antisense transcription of UBE3A-ATS proceed in opposite directions until a transcriptional collision occurs between the RNA polymerases. This physically obstructs the elongation of UBE3A transcription, resulting in almost no functional UBE3A protein being produced from the paternal chromosome.
Consequently, under normal conditions, the maternal UBE3A is virtually the only functional source of the protein in the nervous system. When maternal UBE3A function is lost, there is no compensatory expression left, leading to the hallmark neurological phenotypes of Angelman Syndrome: developmental delay, severe speech impairment, ataxia, seizures, and a characteristic behavioral features.
Mechanisms of Disease
When these parent-specific expression patterns are disrupted, either PWS or AS occurs. The primary causes include:
- Deletion: Deletion of the 15q11-q13 region itself.
- Uniparental Disomy (UPD): Inheriting both copies of the chromosome from a single parent.
- Imprinting Defect / Imprinting Center Variant: Failure to form the normal methylation imprint.
In PWS, the core issue is the loss of function of genes that should be expressed paternally. Therefore, paternal 15q11-q13 deletion, maternal UPD (both copies from the mother), or imprinting defects that prevent paternal imprints from forming can cause PWS.
In contrast, the core issue in AS is the loss of maternal UBE3A function in neurons. Thus, maternal 15q11-q13 deletion, paternal UPD (both copies from the father), imprinting defects, or pathogenic variants within UBE3A itself can lead to Angelman Syndrome.
In summary, PWS and AS are quintessential genomic imprinting disorders. Although they arise from abnormalities in the same chromosomal region, they lead to entirely different disease phenotypes depending on which parent-specific gene expression is lost.
Interpretation is More Than Just Identifying the “Variant Type”
PWS and AS can arise from diverse mechanisms, including CNVs, UPD, imprinting defects, and single-gene variants. Therefore, in actual clinical interpretation, the following questions become critical:
- Is there a deletion in the 15q11-q13 region?
- If a deletion exists, is it of paternal or maternal origin?
- Is there a possibility of UPD (Uniparental Disomy)?
- Is a methylation abnormality suspected?
- Is there a possibility of a UBE3A single-nucleotide variant (SNV)?
- Does the patient’s phenotype align more closely with PWS or AS?
Even within the same genomic region, the interpretation changes entirely based on the parent-of-origin effect and the phenotypic context. In this regard, PWS and AS are not merely “textbook examples” of genetic diseases; they represent the real-world challenges that interpreters face every day. They remind us that variant detection and variant interpretation are two distinct problems.
Phenotype Context Redefines Interpretation
The clinical context is what ultimately drives the interpretation.
For PWS, key clinical clues include neonatal hypotonia followed by hyperphagia, obesity, growth retardation, and decreased muscle mass.
For AS, while early symptoms may be non-specific, developmental delay, severe speech impairment, ataxia, seizures, and characteristic behavioral patterns typically become prominent over time.
Consequently, the discovery of a CNV in the 15q11-q13 region does not automatically lead to a diagnosis of PWS or AS. A robust interpretation must integrate:
- Phenotype
- Onset
- Inheritance
- Methylation pattern
- Parent-of-origin
This process rarely ends with simple filtering. The interpreter must weave together multiple layers of evidence.
Interpretation as “Structured Reasoning”
PWS and AS perfectly illustrate the evolving landscape of modern genetic diagnostics. Interpretation is no longer just a search for a pathogenic variant. It is a comprehensive process of determining, “Does this result explain the patient?” by synthesizing:
- Variant type
- Genomic region
- Parent-of-origin
- Methylation/imprinting mechanism
- Gene function
- Phenotype match
- Inheritance pattern
- Previously reported cases
- Necessity for additional confirmatory testing
Particularly in diseases where the underlying mechanism is paramount—such as imprinting disorders—interpreters must be equipped to view and organize evidence through a structured lens.
GEBRA Expands the Interpreter’s Analytical Space
GEBRA integrates variant prioritization, phenotype matching, ACMG evidence, CNV analysis, and internal evidence review from WES/WGS data into a single, seamless workflow.
For complex conditions like PWS and AS—where understanding the underlying mechanism and phenotypic context is just as vital as variant detection—it is crucial for interpreters to review evidence within a structured framework rather than as scattered data points.
Discover how GEBRA structures clinical reasoning within your actual interpretation workflow.
FAQ
Q1. What is the most significant difference between Prader-Willi Syndrome (PWS) and Angelman Syndrome (AS)?
A. While both are associated with the 15q11-q13 imprinting region, the key difference lies in which parental allele is affected. PWS is primarily caused by the loss of paternal gene expression, whereas AS is driven by the loss of maternal UBE3A function in neurons.
Q2. Why is the parent-of-origin effect critical in variant interpretation?
A. Because the 15q11-q13 region expresses different genes depending on its parental origin. The same deletion or CNV can result in entirely different phenotypes—PWS or AS—depending on whether it is of paternal or maternal origin. Thus, interpretation must account for genomic location alongside parent-of-origin, methylation status, and phenotypic context.
Q3. Why is UBE3A so important in Angelman Syndrome?
A. In the nervous system, UBE3A is functionally expressed primarily from the maternal allele, while the paternal allele is silenced by an antisense transcript (UBE3A-ATS). If the maternal UBE3A function is lost, there is no functional backup in the neurons, leading to the developmental delays, seizures, and ataxia characteristic of AS.
Q4. How does GEBRA assist in interpreting imprinting disorders like PWS and AS?
A. GEBRA connects variant prioritization, phenotype matching, ACMG evidence, and CNV analysis within a single workflow. This allows interpreters to examine complex genetic evidence through a structured interface, facilitating more accurate and efficient reasoning.
References
- Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greenswag LR, Whitman BY, Greenberg F. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics. 1993;91(2):398-402. PMID: 8424017; PMCID: PMC6714046.
- Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14(1):10-26. doi:10.1038/gim.0b013e31822bead0.
- Driscoll DJ, Miller JL, Schwartz S, Cassidy SB. Prader-Willi Syndrome. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle GeneReviews; 1998–2025. Updated 2024 Mar 14. Available from: NCBI Bookshelf NBK1144
- Roberts CT, Arezoumand KS, Kadar Shahib A, Davie JR, Rastegar M. Epigenetics in rare neurological diseases. Front Cell Dev Biol. 2024;12:1413248. doi:10.3389/fcell.2024.1413248. PMID: 39108836; PMCID: PMC11300358
. - Ma VK, Mao R, Toth JN, Fulmer ML, Egense AS, Shankar SP. Prader-Willi and Angelman syndromes: mechanisms and management. Appl Clin Genet. 2023;16:41-52. doi:10.2147/TACG.S372708. PMID: 37051256; PMCID: PMC10084876.
- Mapendano CK, Kishino T, Miyazaki K, Kondo S, Yoshiura KI, Hishikawa Y, Koji T, Niikawa N, Ohta T. Expression of the Snurf-Snrpn IC transcript in the oocyte and its putative role in the imprinting establishment of the mouse 7C imprinting domain. J Hum Genet. 2006;51(3):236-243. doi:10.1007/s10038-005-0351-8. PMID: 16429232.
- Groh M, Gromak N. Out of balance: R-loops in human disease. PLoS Genet. 2014;10(9):e1004630. doi:10.1371/journal.pgen.1004630. PMID: 25233079; PMCID: PMC4169248.
- Mabb AM, Judson MC, Zylka MJ, Philpot BD. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011;34(6):293-303. doi:10.1016/j.tins.2011.04.001. PMID: 21592595; PMCID: PMC3116240.
- McCann KL, Baserga SJ. Long noncoding RNAs as sinks in Prader-Willi syndrome. Mol Cell. 2012;48(2):155-157. doi:10.1016/j.molcel.2012.10.005. PMID: 23102265; PMCID: PMC3496270.
- Polex-Wolf J, Lam BY, Larder R, Tadross J, Rimmington D, Bosch F, et al. Hypothalamic loss of Snord116 recapitulates the hyperphagia of Prader-Willi syndrome. J Clin Invest. 2018;128(3):960-969. doi:10.1172/JCI97007. PMID: 29376887; PMCID: PMC5824864.
- Butler MG, Silvey S, van Bosse HJP. Barriers, limitations, and experiences with clinical trials-treatment in rare diseases with Prader-Willi syndrome as an example. Genes (Basel). 2025;16(12):1436. doi:10.3390/genes16121436. PMID: 41465109; PMCID: PMC12732491.
- Milazzo C, Mientjes EJ, Wallaard I, Rasmussen SV, Erichsen KD, Kakunuri T, et al. Antisense oligonucleotide treatment rescues UBE3A expression and multiple phenotypes of an Angelman syndrome mouse model. JCI Insight. 2021;6(15):e145991. doi:10.1172/jci.insight.145991. PMID: 34369389; PMCID: PMC8410092.
Get exclusive rare disease updates
from 3billion.

3billion Inc.
3billion is dedicated to creating a world where patients with rare diseases are not neglected in diagnosis and treatment.