마르판 증후군: 유전적 원인과 Ghent 진단 기준 요약
📍Key Takeaways
- 마르판 증후군의 약 90%는 FBN1 병원성 변이에 의해 발생하며, TGFBR1/2 및 FBN2 변이는 유사한 표현형을 나타낼 수 있습니다.
- 진단은 개정 Ghent Nosology(2010, 마르판 증후군을 판별하는 세계 공통의 진단 표준)에 따르며, 대동맥근 확장과 수정체 탈구(ectopia lentis)가 핵심 기준입니다.
- 대동맥 박리(주로 Type A)는 주요 사망 원인으로, 정기적인 심초음파 감시가 필수적입니다.
- 임상 기준이 불명확하거나, 가족 검사 또는 자녀 계획이 필요한 경우 유전자 검사가 적극 권장됩니다.
- 유전자형-표현형 상관관계는 대동맥 위험 층화 및 수술 시점 결정에 실질적인 임상 의미를 갖습니다.

마르판 증후군(Marfan syndrome)은 FBN1유전자의 병원성 변이로 인해 발생하는 전신성 결합 조직 질환입니다. 전 세계적으로 약 5,000명 중 1명의 유병률을 보이며, 조기에 진단하고 적절히 관리하면 충분히 예방 가능한 심혈관 합병증을 동반합니다. 하지만 문제는 확실한 진단을 받기까지 걸리는 시간입니다. 문헌에 보고된 평균 진단 지연은 5~7년에 달하며, 이는 여전히 해결되지 않은 과제입니다.
본 리뷰는 마르판 증후군이 의심되는 환자를 진료하는 임상의가 진단부터 유전 검사, 관리까지 한눈에 파악하고 실행할 수 있도록 핵심 체계를 정리한 근거 기반 참고 자료입니다.
1. 마르판 증후군이란?
마르판 증후군은 심혈관계, 근골격계, 안과 계통에 영향을 미치는 상염색체 우성 결합 조직 질환입니다. 1896년 Antoine Marfan이 처음 기술하였으며, 분자적 기전 (FBN1 유전자의 병원성 변이에 의한 피브릴린-1(fibrillin-1) 단백 이상) 은 1991년 Dietz 등에 의해 규명되었습니다.
전 세계 유병률은 5,000~10,000명 중 1명으로 추정되며, 성별 또는 인종적 편향은 없습니다. 약 75%는 부모로부터 유전되며, 나머지 25%는 de novo 변이로 발생합니다. 유전자를 가졌다면 증상은 반드시 나타나지만, 그 정도나 부위는 같은 가족이라도 사람마다 제각각 다릅니다.

2. 유전적 기전: FBN1과 그 너머
FBN1 유전자는 15번 염색체(15q21.1)에 있으며, 세포 사이를 채우는 지지 구조인 ‘세포외기질’이 튼튼하게 유지되도록 돕는 ‘피브릴린-1’ 단백질을 만듭니다. 현재까지 3,000개 이상의 병원성 변이가 보고되었으며, 미스센스 변이(유전자 정보 중 하나가 바뀌어 단백질의 구성 성분인 아미노산 종류가 달라지는 현상)가 가장 흔합니다.
피브릴린-1 기능 이상은 두 가지 효과를 초래합니다.
- 구조적 취약화: 탄성 조직(대동맥, 수정체 소대, 골막)의 약화
- TGF-β 신호 전달 이상: 대동맥 벽 리모델링의 핵심 기전 (주요 치료 표적)
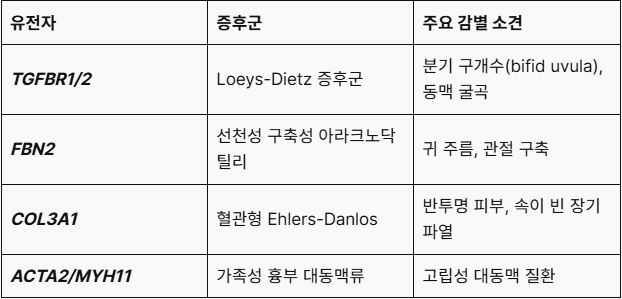
[FBN1 변이가 아닐 때 의심해 봐야 할 질환]
3. 계통별 임상 특징
1) 심혈관계 (생명 위협)
- 대동맥근 확장/동맥류: 성인의 약 80%에서 관찰
- 대동맥 박리: Valsalva동 수준의 Type A
- 승모판 탈출증(MVP): 약 75%, 역류 위험 동반
- 폐동맥 확장
2) 근골격계
- 사지 불균형적 장골(dolichostenomelia)을 동반한 큰 키
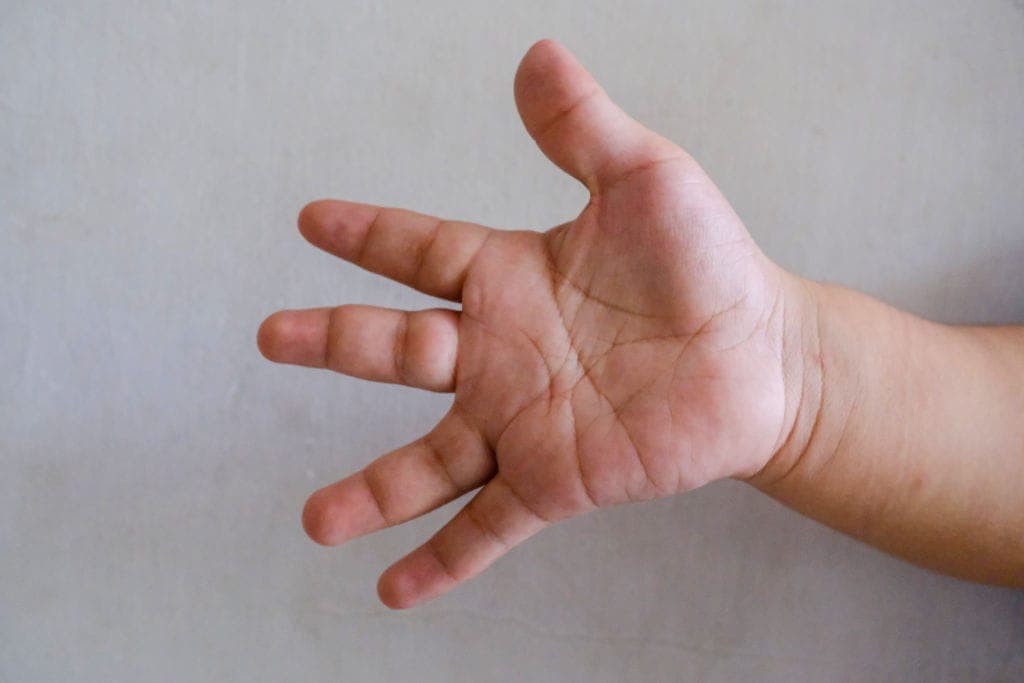
- 거미손가락(arachnodactyly): Steinberg 엄지 징후, Walker-Murdoch 손목 징후 양성
- 오목가슴(pectus excavatum) 또는 새가슴(carinatum)
- 척추 측만증 (중증의 경우 >40°)
3) 안과
- 수정체 탈구(ectopia lentis): 환자의 약 60%에서 발생; 상측두방향 탈위가 특징적
- 심한 근시 (>3디옵터) 및 망막 박리 위험 증가
4) 기타
- 자발성 기흉 (약 10%)
- 경막 확장증(dural ectasia): 60% 이상에서 관찰 (만성 요통의 원인)
4. 진단 기준: Ghent Nosology
2010년 개정된 Ghent Nosology 기준은 대동맥근 확장과 수정체 탈구를 핵심 지표로 강화하고, 비특이적 골격 소견의 비중을 낮췄습니다.
[전신 점수 (Systemic Score)] – ≥7점 시 의미 있음
| 소견 | 점수 |
| 손목 + 엄지 징후 모두 양성 | 3 |
| 손목 또는 엄지 징후 중 하나만 양성 | 1 |
| 새가슴 | 2 |
| 오목가슴 또는 흉벽 비대칭 | 1 |
| 뒤꿈치 변형 | 2 |
| 단순 평발(flat pes planus) | 1 |
| 자발성 기흉 | 2 |
| 경막 확장증 | 2 |
| 비구 돌출증 | 2 |
| 상하체 비율 감소 + 팔 길이/키 비율 증가 | 1 |
| 척추 측만증 또는 흉요추 후만 | 1 |
| 팔꿈치 신전 제한 | 1 |
| 안면 특징 (5개 중 3개) | 1 |
| 피부 선조 | 1 |
| 근시 >3D | 1 |
| 승모판 탈출증 | 1 |
※ Ghent Nosology 기반, 다음 중 하나를 충족하면 마르판 증후군으로 의심 권고:
- 대동맥근 확장(Z-score ≥2) + 수정체 탈구
- 대동맥근 확장 + FBN1 병원성 변이
- 대동맥근 확장 + 전신 점수 ≥7
- 수정체 탈구 + 대동맥 확장과 관련된 것으로 알려진 FBN1 변이

5. 유전자 검사가 필요한 경우
다음 상황에서는 유전자 검사가 강력히 권장됩니다.
- 임상 기준 불명확: 전신 점수 경계치이거나 진단을 위한 결정적 증상이 부족한 경우
- 소아 및 청소년: 임상 소견이 아직 완성되지 않은 시기의 조기 진단
- 가족 연쇄 검사: 가족 중 이미 변이가 확인된 환자가 있어, 다른 가족의 유전 여부를 확인할 경우
- 비전형적 표현형: 로이스-디츠 증후군 등 증상이 비슷한 다른 결합 조직 질환과 구분이 필요한 경우
- 자녀 계획: 착상 전 유전자 검사(PGT) 및 산전 진단을 원하는 경우
6. 관리 및 모니터링
- 심혈관계: 대동맥 근부 지름이 4.5cm 미만이면 매년, 4.5cm 이상이거나 확장 속도가 빠르면 6개월 간격으로 시행합니다.
- 생활 습관: 경쟁적/접촉 스포츠 및 등척성 운동(무거운 것 들기 등) 금지.
- 임신: 대동맥근 >4.0cm 시 임신에 따른 위험 크므로 다학제적 관리 필수
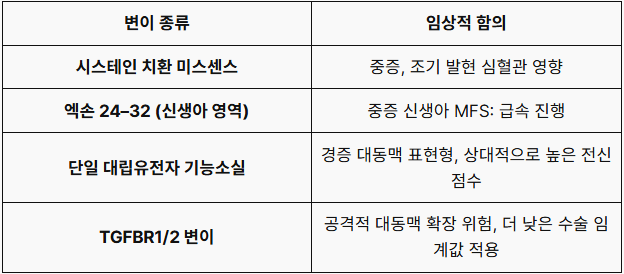
7. 유전자형-표현형 상관관계
8. 결론
마르판 증후군은 예방 가능한 조기 대동맥 박리의 원인이 됩니다. 치료의 핵심은 조기 정확 진단과 지속적 모니터링입니다. 현재, 유전자 검사는 진단 확정을 넘어 변이 특이적 예후 예측과 가족 관리를 위한 필수적인 도구로 자리 잡았습니다.
복잡한 감별 진단, 3billion의 유전자 검사가 답을 찾아드립니다. 아래 버튼을 눌러 3billion의 WES, WGS 검사에 대해 문의해 보세요.
References
- Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476-485. https://doi.org/10.1136/jmg.2009.072785
- Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337-339. https://doi.org/10.1038/352337a0
- Judge DP, Dietz HC. Marfan’s syndrome. Lancet. 2005;366(9501):1965-1976. https://doi.org/10.1016/S0140-6736(05)67789-6
- Pyeritz RE. Marfan syndrome: improved clinical history results in expanded natural history. Genet Med. 2019;21(8):1683-1690. https://doi.org/10.1038/s41436-018-0399-4
- Faivre L, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations. Am J Hum Genet. 2007;81(3):454-66. https://doi.org/10.1086/520125
- Isselbacher EM, Ouriel K, Silberbach M, et al. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2022;80(24):e223-e393. https://doi.org/10.1016/j.jacc.2022.08.004
3billion 뉴스레터 구독자만을 위한
희귀질환 진단 최신 정보를 받아보세요.

Soo-jung Baek
희귀질환 진단이라는 막막한 길 위에서, 정확한 정보와 따뜻한 공감으로 여러분의 여정에 끝까지 함께하고 싶은 마케터 백수정입니다.