What’s New in CHARGE Syndrome Research: A 2024–2026 Update
📍Key Takeaways
- CHARGE syndrome is a rare congenital disorder most commonly caused by pathogenic CHD7 variants, presenting with anomalies of the eyes, heart, choanae, growth, genitalia, and ears.
- Hearing loss affects 97% of patients; nonsense/frameshift variants carry the highest SNHL risk. Cochlear implant outcomes are poorer than in other neurocristopathies, though meaningful auditory gains remain achievable.
- Olfactory dysfunction exceeds 80% prevalence yet is absent from current Blake/Verloes criteria — its links to neurodevelopmental delay and reduced quality of life make it a strong candidate for future diagnostic revisions.
- Coronal clival cleft on fetal MRI and olfactory sulcus abnormalities on neurosonography have been proposed as emerging prenatal markers that may prompt earlier CHD7 testing.
- Growing adult cohorts show high rates of anxiety, OCD, sleep disturbances, and chronic pain, highlighting the need for structured transition care beyond the pediatric setting.
A Well-Characterized Syndrome With Much Still to Learn
CHARGE syndrome is a rare, complex congenital disorder most commonly caused by pathogenic variants in the CHD7 gene. The acronym reflects its cardinal features: Coloboma, Heart defect, Atresia of the choanae, Retardation of growth, Genital hypoplasia, and Ear anomaly/deafness.
CHD7 was identified as the causative gene in 2004. Two decades later, research into CHARGE syndrome remains highly active. As survival rates have improved, an adult patient cohort has begun to emerge, bringing long-term outcomes and mental health into focus as new areas of inquiry.
Between 2024 and 2026, the research landscape surrounding CHARGE syndrome has shifted considerably. A series of notable publications has addressed hearing, olfaction, prenatal imaging, immunity, endocrinology, and mental health. This post summarizes the key developments.
1. Hearing Loss — Variant Type Shapes Prognosis
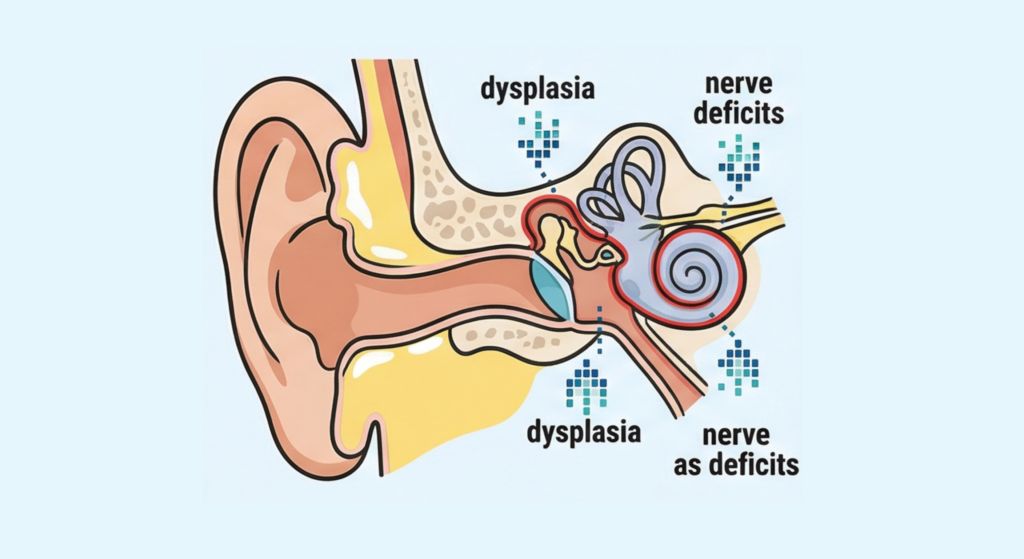
Hearing loss is among the most consistent findings in CHARGE syndrome, yet recent data suggest its prevalence and severity may be greater than previously recognized.
A cohort study conducted at Cincinnati Children’s Hospital (n=57) examined the relationship between CHD7 variant type and the nature of hearing loss in pediatric patients. Hearing loss was identified in 97% of the cohort, and 83% demonstrated severe-to-profound loss in at least one ear — figures exceeding those reported in prior literature. A statistically significant association was observed between variant type and hearing loss classification (p=0.002), with haploinsufficiency resulting from nonsense or frameshift variants conferring the greatest risk for sensorineural hearing loss (SNHL). The authors also emphasized the importance of systematic evaluation, including temporal bone CT, for accurate prevalence estimates. ¹
A separate study compared cochlear implantation (CI) outcomes across neurocristopathies, including Waardenburg, Noonan, and Kabuki syndromes. Patients with CHARGE syndrome demonstrated markedly poorer auditory and speech outcomes relative to the other groups, attributed to CHD7-related cochlear nerve dysplasia and central auditory deficits. Age at implantation and neurodevelopmental level as assessed by the Gesell Developmental Schedules were identified as independent predictors of post-CI auditory performance. ² Notably, a separate CHARGE-specific CI series reported meaningful auditory gains following implantation, with approximately 43% of children achieving functional speech perception despite universal cochlear nerve deficiency — suggesting that, while outcomes are comparatively limited, cochlear implantation can still confer clinically significant benefit in appropriately selected patients. ²
Also in 2026, a study systematically evaluated cochlear angular orientation in patients with CHARGE syndrome, proposing a new consideration for preoperative imaging protocols. ³
2. Olfactory Dysfunction — At the Center of Diagnostic Criteria Debates
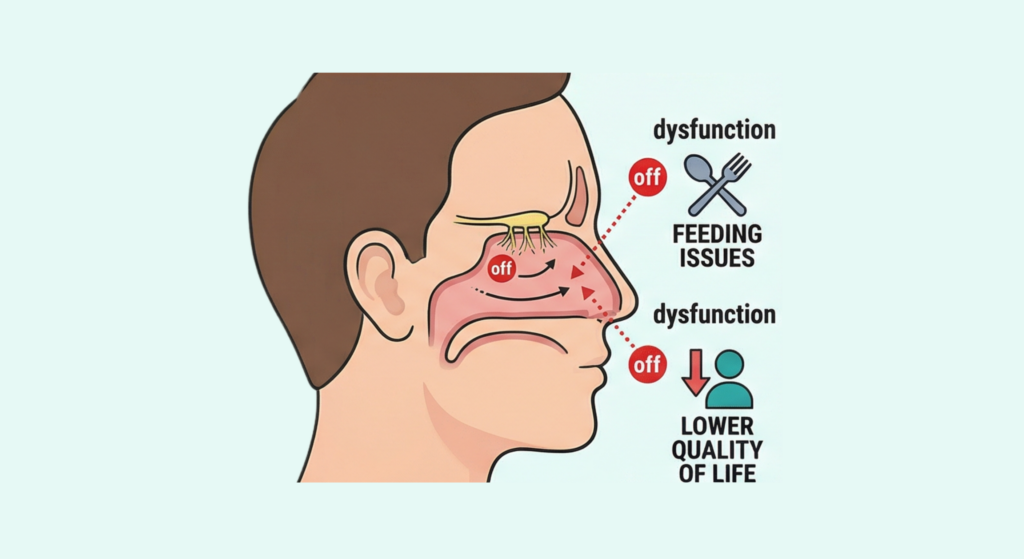
Olfactory dysfunction (OD) has long been an underrecognized feature of CHARGE syndrome. A 2025 systematic review addressed this gap directly.
Registered with PROSPERO, the review screened 1,643 records across six databases, ultimately analyzing 16 studies. The central finding was that OD prevalence exceeds 80% in CHARGE syndrome — yet it is not included in the current Blake/Verloes diagnostic criteria. Radiologic imaging remains the most commonly used assessment modality; psychophysical testing and caregiver report are infrequently employed. The authors presented data linking OD to neurodevelopmental delays, feeding and swallowing difficulties, and reduced quality of life, and called for the development of pediatric-specific, developmentally appropriate olfactory assessment tools. One of the review’s conclusions was that OD warrants inclusion in any future revision of the CHARGE diagnostic criteria. ⁴
3. Prenatal Diagnosis — New Imaging Markers Emerging
Prenatal diagnosis of CHARGE syndrome is valuable for timely genetic counseling and delivery planning, yet data on fetal neuroimaging findings have been limited. Two recent publications have begun to address this gap.
The first, a retrospective study from Children’s Hospital of Philadelphia (CHOP), analyzed fetal MRI findings in 18 genetically confirmed cases of CHARGE syndrome. A coronal clival cleft was identified in 72% of cases and confirmed on postnatal imaging in all cases where it was available — a finding not previously well characterized as a prenatal imaging marker for CHARGE. Additional findings included inner ear dysplasia (identified in all evaluable cases), olfactory apparatus hypoplasia (83%), ocular dysmorphia or coloboma (44%), choanal atresia (39%), and cerebellar malformation (17%). ⁵
The second study prospectively evaluated 147 fetuses with congenital heart defects, assessing olfactory sulcus (OS) development as part of routine neurosonography. Among the four cases with abnormal OS findings, CHD7 variants were confirmed in three. No cases with normal OS findings were subsequently diagnosed with CHARGE syndrome postnatally. ⁶
Both studies suggest that identification of these findings on prenatal imaging may help shorten the interval to genetic confirmation.
4. CHARGE Syndrome in Adults — What Becomes Visible as Survivors Age

The historically high infant mortality associated with CHARGE syndrome long precluded meaningful study of long-term outcomes. As survival rates have improved, an adult cohort has emerged, and with it, clinical questions that fall outside the scope of pediatric-centered research.
A study published in the Journal of Neurodevelopmental Disorders in 2026 examined the prevalence and clinical characteristics of anxiety disorders and obsessive-compulsive disorder (OCD) in adults with CHARGE syndrome, with additional analysis of pain and sleep disturbances. The findings underscore that the behavioral phenotype of CHARGE syndrome extends well beyond childhood and requires ongoing management into adulthood, supporting the development of structured transition care pathways. ¹¹
How is CHARGE syndrome diagnosed in real clinical settings?
3billion holds data on 91 patients confirmed with CHARGE syndrome through WES/WGS (as of March 2026). Multiple symptoms are recorded across each case, making simple aggregation meaningless — but it is possible to examine which symptom categories appeared across how many patients.
Looking at the symptoms present among these 91 patients, the most broadly distributed were neurodevelopmental delay and hypotonia (~63%) and hearing/ear anomalies (~62%), both confirmed in well over half of all patients. Cardiac malformations (~51%) and craniofacial anomalies (~49%) were also co-occurring in a large proportion of patients. Genital anomalies (micropenis, cryptorchidism, etc., ~36%) and endocrine findings (thyroid, pituitary abnormalities, etc., ~21%) were likewise recorded in a notable share of patients — consistent with the IHH/Kallmann syndrome overlap and congenital hyperinsulinism cases discussed elsewhere in this article.
In contrast, choanal atresia — one of the hallmark diagnostic criteria for CHARGE — was identified in only ~5% of patients, and ocular coloboma in just ~14%.
The timing of symptom onset also varied considerably. Some cases had abnormalities documented as early as the antenatal period, while others presented symptoms only during childhood or adolescence. The real-world data make clear that CHARGE syndrome is not a condition caught solely in the neonatal period.
As the studies reviewed here demonstrate, CHARGE syndrome presents a broad and heterogeneous phenotypic spectrum. Single-symptom presentations are common, and the diagnostic pathway varies considerably from case to case. Variant types that may be missed by single-gene testing or limited gene panels — including deep intronic splice variants and mosaic changes — are increasingly recognized as clinically relevant. The role of WES and WGS in establishing a diagnosis and informing management continues to grow.
For cases where CHARGE syndrome is suspected or differential diagnosis with an overlapping syndrome is needed, please feel free to inquire about 3billion’s WES/WGS testing, offered with automated reanalysis technology.
References
All references cited in this post are peer-reviewed articles published between 2024 and 2026.
[1] Kettler M, Simpson B, Meinzen-Derr J, et al. Genetic and Clinical Predictors of Hearing Loss Among Patients with CHARGE Syndrome. J Am Acad Audiol. 2025;36(1):45–52. → DOI: 10.3766/jaaa.230055 | PMID: 40015694 → Cited in: Section 1 (hearing loss prevalence; genotype-phenotype correlation)
[2] Ni K, Fang J, Fan W, et al. Analysis of the clinical features of neurocristopathy-related hearing loss and how these relate to outcomes after cochlear implantation. Sci Rep. 2025;15(1):41227. → DOI: 10.1038/s41598-025-25126-w | PMID: 41271950 → Cited in: Section 1 (predictors of cochlear implantation outcomes; CHD7-related cochlear nerve dysplasia)
[3] Eroğlu E, Küleçi Ç, Aydın C, Sennaro ğlu L. Evaluation of cochlear angular orientation in patients with CHARGE syndrome. Eur Arch Otorhinolaryngol. 2026. → DOI: 10.1007/s00405-026-10195-y | PMID: 41862720 → Cited in: Section 1 (cochlear angular orientation abnormalities; preoperative imaging protocol)
[4] Spencer GM, Karim K, Coyle P, Bhargava EK, Whitcroft KL. Olfactory dysfunction in CHARGE syndrome: a systematic review of prevalence, assessment methods, and clinical correlates. Rhinology. 2025. → DOI: 10.4193/Rhin25.391 | PMID: 41313596 → Cited in: Section 2 (olfactory dysfunction prevalence >80%; inclusion in diagnostic criteria)
[5] Teixeira SR, Cerron-Vela C, Khalek N, Wright R, Whitehead MT. Coronal Clival Cleft in CHARGE Syndrome: Fetal MRI Series. AJNR Am J Neuroradiol. 2025;46(5):1022–1028. → DOI: 10.3174/ajnr.A8609 | PMID: 40316448 → Cited in: Section 3 (coronal clival cleft; fetal MRI findings spectrum)
[6] Charach R, Pérez-Cruz M, Masoller N, et al. Systematic Ultrasound Evaluation of Olfactory Sulci in Fetuses with Congenital Heart Defects: A Clue for CHARGE Syndrome Diagnosis. Fetal Diagn Ther. 2025;52(3):280–290. → DOI: 10.1159/000543190 | PMID: 39809240 → Cited in: Section 3 (olfactory sulcus assessment; contribution to prenatal CHARGE diagnosis via neurosonography)
[7] Zhao J, Hu R, Lai KC, et al. Generation of induced pluripotent stem cells from a patient with CHARGE syndrome with athymia, harboring a heterozygous mutation in CHD7. Stem Cell Res. 2026;91:103912. → DOI: 10.1016/j.scr.2026.103912 | PMID: 41539084 → Cited in: Section 4 (iPSC generation; research platform for thymopoiesis)
[8] Ibeas C, Giraudo F, Männistö JME, Flanagan SE, Mericq V. Congenital hyperinsulinism in an individual with CHARGE syndrome and a pathogenic CHD7 variant. JCEM Case Rep. 2026;4(3):luag016. → DOI: 10.1210/jcemcr/luag016 | PMID: 41743177 → Cited in: Section 5-1 (CHARGE syndrome with congenital hyperinsulinism; diazoxide response)
[9] Wu J, Huang Z, Zhu B, et al. De novo CHD7 variant in a CHARGE syndrome preterm infant initially diagnosed as idiopathic hypogonadotropic hypogonadism: a case report and literature review. BMC Pediatr. 2025;25(1):926. → DOI: 10.1186/s12887-025-06251-x | PMID: 41225453 → Cited in: Section 5-2 (initial diagnosis of IHH; CHD7 variant identified via WES; CHARGE in a preterm infant)
[10] Wang Y, Zhai J, Habibi I, et al. Clinical and genetic basis of congenital gonadotropin deficiency. Hum Reprod Open. 2026;2026(2):hoag017. → DOI: 10.1093/hropen/hoag017 | PMID: 41873429 → Cited in: Section 5-2 (cohort of 568 patients; genetic overlap in syndromic hypogonadotropic hypogonadism)
[11] Madhavan-Brown SA, Hartshorne TS, Schneider SB, Slavin LJ. Anxiety and obsessive-compulsive disorder (OCD) in adults with CHARGE syndrome. J Neurodev Disord. 2026. → DOI: 10.1186/s11689-026-09673-5 | PMID: 41765890 → Cited in: Section 6 (anxiety and OCD in adult CHARGE patients; behavioral phenotype in adulthood)
Get exclusive rare disease updates
from 3billion.

Soo-jung Baek
As a marketer, I strive to empower the rare disease community by sharing meaningful insights backed by our company’s expertise.