Cleidocranial Dysplasia: Why Genetic Testing Matters and When to Consider It
📍 Key Takeaways
- CCD is an autosomal dominant rare skeletal dysplasia caused by RUNX2 variants (prevalence ~1/1,000,000)
- Major phenotypes: supernumerary teeth/eruption failure (93.5%), clavicular anomalies (84.3%), delayed fontanelle closure, midface hypoplasia
- 72% of variants are de novo → genetic testing is essential even without family history when characteristic features are present
- Testing strategy: single-gene/panel testing first; expand to WES·CNV analysis (MLPA/WGS) if negative
- Early diagnosis is key to preventing dental complications and enabling cascade testing within families
1. What is CCD: Core Clinical Phenotypes
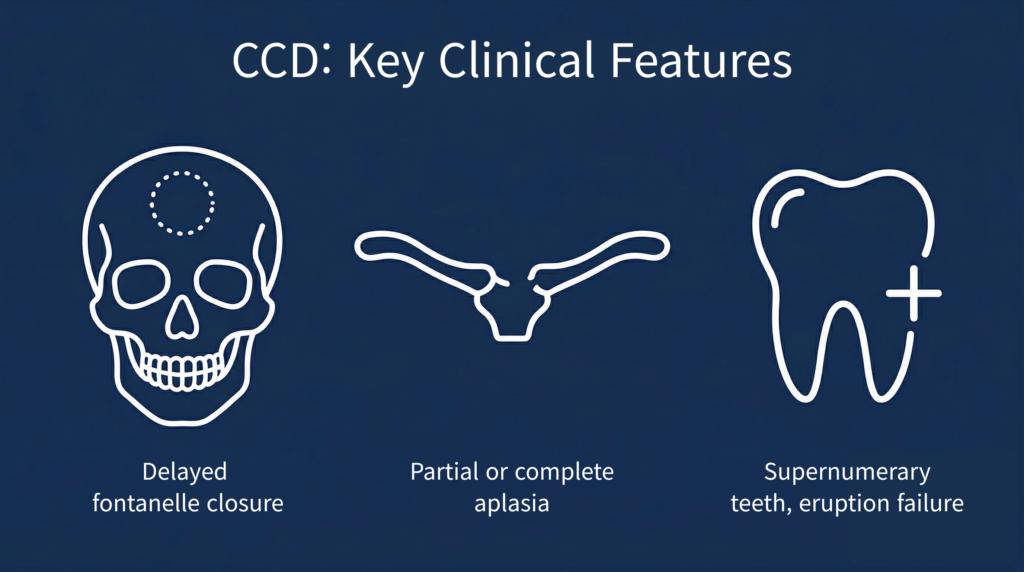
Cleidocranial Dysplasia (CCD) is an autosomal dominant rare skeletal dysplasia caused by RUNX2 gene variants, with a reported birth prevalence of approximately 1/1,000,000.[1]
A systematic review of 283 CCD patients found dental anomalies — including supernumerary teeth and permanent tooth eruption failure — in over 93.5% of patients, and skeletal anomalies such as clavicular abnormalities and maxillary hypoplasia in over 84.3%.[2] A Turkish cohort of 51 CCD patients confirmed delayed closure of the anterior fontanelle in 89%, midface hypoplasia in 94%, and abnormal shoulder mobility in 90%.[3]
2. RUNX2 Gene and Variant Spectrum

RUNX2 is located at 6p21.1 and encodes a transcription factor specifically expressed in osteoblast differentiation.[4] A recent systematic review (2024) analyzing 569 RUNX2 variants from 103 publications and 453 CCD patients found that variants consist of 48.68% in-frame variants and 51.32% null variants, with the RHD (Runt homology domain) being the most affected functional region at 55.54%.[5] In the Turkish cohort, small sequence variants accounted for 90% and whole-gene deletions for 10%.[3]
3. Genotype-Phenotype Correlations
Phenotypic variability exists even among family members carrying the same variant, likely attributable to genetic modifiers and hypomorphic effects.[6] However, a 2024 systematic review reported statistically significant associations between variant location and specific phenotypes. Missense variants within the RHD were significantly associated with supernumerary teeth, macrocephaly, delayed fontanelle closure, hypertelorism, and limited shoulder abduction, while in-frame insertions/deletions tended to be associated with fewer CCD phenotypes compared to other variant types.[5]
4. When to Consider Genetic Testing
Since de novo variants account for 72.0% of CCD cases, genetic testing should be considered even in the absence of family history when the following features are identified:[2]
- Partial or complete absence of the clavicles (confirmed by X-ray)
- Delayed closure of the anterior fontanelle (persistent opening after birth)
- Combined findings of supernumerary teeth and delayed primary tooth shedding
- Characteristic craniofacial features with midface hypoplasia
- Family history of the above findings (autosomal dominant)
5. Genetic Testing Strategy
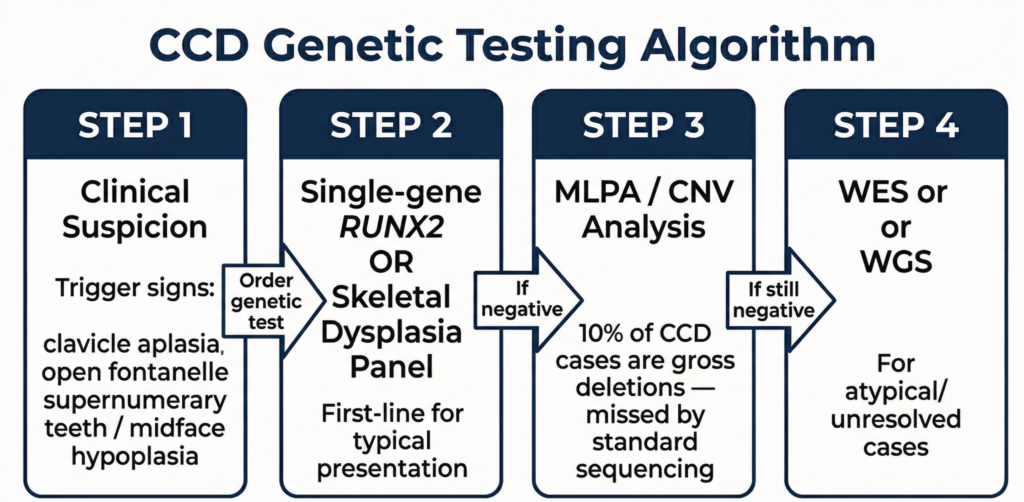
Cases have been reported in which novel RUNX2 variants were identified by WES (whole-exome sequencing) in patients clinically suspected of CCD but whose diagnosis had been delayed by standard sequencing.[7] This suggests that while single-gene testing or a skeletal dysplasia panel is recommended as the first-line approach for typical phenotypes, the strategy should be expanded to WES or CNV analysis when results are negative. In particular, since whole-gene deletions may not be detected by standard sequencing,[3] MLPA or WGS-based CNV analysis should be considered in parallel when clinical suspicion is high.
How is CCD diagnosed in real clinical settings? 3billion analyzed data from 21 patients diagnosed with CCD through genetic testing (as of March 2026).
Test Results: SNVs accounted for the majority of variant types at ~81%, followed by CNVs at ~14% and expansions (EXP) at ~5%.
Age of Onset: The neonatal period was the most common time of presentation.
Key Clinical Phenotypes: Looking at the symptoms present among these 21 patients, delayed closure of the anterior fontanelle/wide fontanelle was the most frequently observed finding, followed by supernumerary teeth and delayed tooth eruption, clavicular hypoplasia or aplasia, short stature, and craniofacial dysmorphism (broad forehead, hypertelorism, and depressed nasal bridge). These findings are consistent with the core CCD phenotypes reported in the literature.
Notable Case: One case of parietal foramina with cleidocranial dysplasia caused by an MSX2 variant — rather than the typical RUNX2 — was included, highlighting the need for differential genetic testing in patients presenting with similar phenotypes.
Early diagnosis of CCD is directly linked to the prevention of dental complications and improvement of quality of life.[1] Genetic counseling should address both the autosomal dominant inheritance pattern and the high rate of de novo variants.[2] Early genetic diagnosis enables timely establishment of an appropriate management plan and cascade testing within the family, and the clinical value of genetic testing in diagnosing cleidocranial dysplasia continues to grow.
Considering WES/WGS testing? Contact 3billion for details on cost, process, and required materials. Our specialists are ready to help.
References
- Inchingolo AD, et al., Cleidocranial Dysplasia: Etiopathogenesis, Diagnosis, and Therapeutic Approach (2021) https://doi.org/10.3390/medicina57121350
- Golan I, et al., Cleidocranial dysplasia: clinical and radiological manifestations (2003) https://doi.org/10.1259/dmfr/63490079
- Berkay EG, et al., Clinical and molecular characterization of cleidocranial dysplasia patients from Turkey (2021) https://doi.org/10.1002/ajmg.a.62261
- Otto F, et al., Cbfa1-related disorders: mutations, phenotype, and biological implications (2002) https://doi.org/10.1002/humu.10043
- Thaweesapphithak S, et al., Comprehensive analysis of RUNX2 variants and genotype-phenotype correlations in cleidocranial dysplasia (2024) https://doi.org/10.1186/s12967-024-05904-2
- Zhang C, et al., Variable expressivity of a novel heterozygous RUNX2 mutation in a Chinese family with cleidocranial dysplasia (2010) https://doi.org/10.1093/mutage/geq044
- Ma D, et al., A novel RUNX2 mutation identified by whole-exome sequencing in a patient with cleidocranial dysplasia (2018) https://doi.org/10.1097/MD.0000000000011328
Get exclusive rare disease updates
from 3billion.

Soo-jung Baek
As a marketer, I strive to empower the rare disease community by sharing meaningful insights backed by our company’s expertise.





