Genetic Variants Determine Prognosis: Genotype–Phenotype Correlations in Congenital and Infantile Nephrotic Syndrome
📍 Key Takeaways
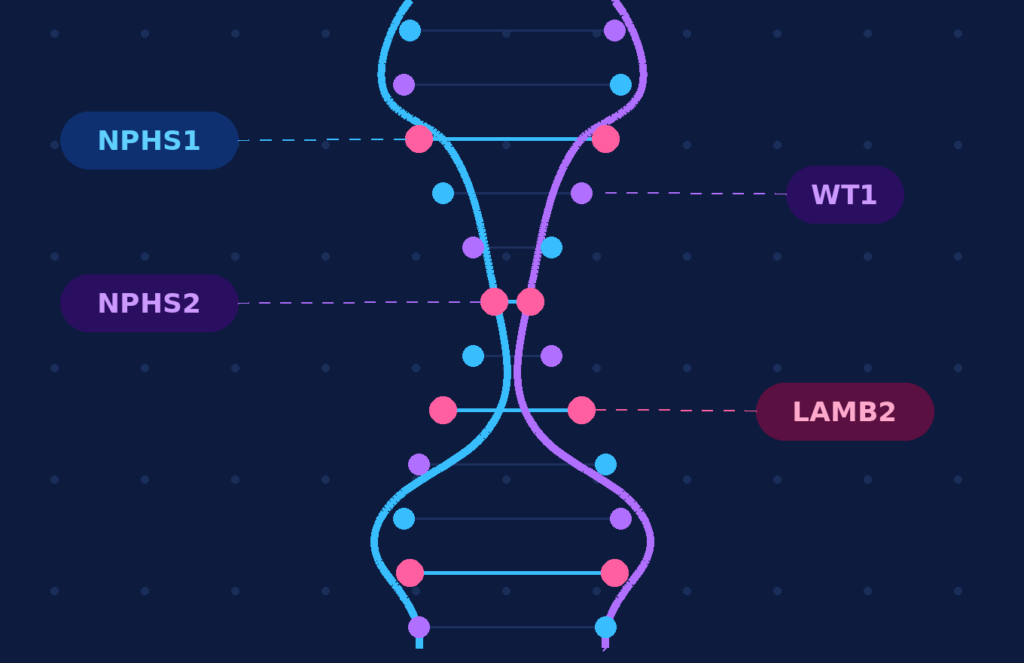
• More than 50% of CNS and infantile NS cases have a monogenic cause; four genes — NPHS1, NPHS2, WT1, and LAMB2 — account for two-thirds of all cases.
• Even within the same gene, earlier onset (CNS) is associated with a more severe genotype and faster progression to renal failure.
• Compound heterozygous NPHS1 variants, and digenic variants involving NPHS1/WT1 or NPHS1/NPHS2, are strongly associated with the need for transplantation.
• Without genetic testing results, systematic prediction of steroid responsiveness, transplant planning, and family counseling cannot be performed.
• A “negative single-gene test” does not rule out a genetic etiology; comprehensive panels or WES should be considered.
Same Diagnosis, Different Prognosis
Congenital nephrotic syndrome (CNS, onset at 0–3 months of age) and infantile nephrotic syndrome (INS, onset at 3–12 months) may appear clinically similar. However, the causative gene and variant type fundamentally determine the timing of renal failure, the need for transplantation, and whether pretransplant nephrectomy should be considered.
This article reviews current evidence to summarize phenotypic characteristics by major causative gene and discusses why genotypic information should serve as the starting point for clinical decision-making.
─────────────────────────────
- Genetic Architecture of CNS and Infantile NS
A significant proportion of patients with CNS or infantile NS have a monogenic cause. In a classic European cohort, 66.3% of patients with onset within the first year of life were explained by four genes: NPHS1, NPHS2, WT1, and LAMB2. A causative variant was identified in 84.8% of CNS cases and 44.1% of infantile NS cases.
In this cohort, NPHS2 was the most common causative gene in both groups, while NPHS1 variants were identified exclusively in congenital-onset cases.
Table 1. Distribution of Major Causative Genes by Age of Onset¹
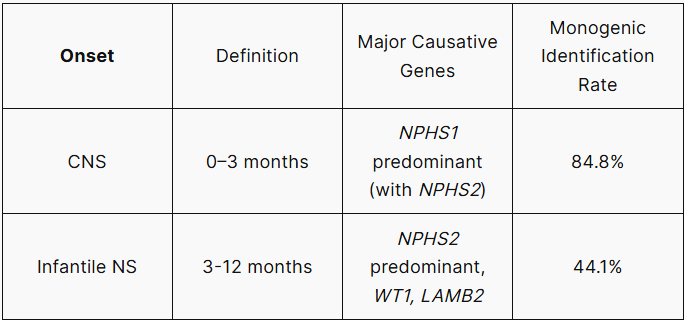
Note: NPHS1 variants are identified almost exclusively in CNS and are rarely observed in infantile NS.¹
─────────────────────────────
- Phenotypic Characteristics by Gene
2-1. NPHS1 (Nephrin): The Core Determinant of Prognosis
NPHS1 encodes nephrin, the key structural protein of the slit diaphragm. In a Japanese multicenter study (n=74), patients with NPHS1 variants reached renal failure significantly later than those with variants in other genes (median 31.0 months vs. 1.0 months in the CNS group; p<0.001).² However, even within NPHS1 variant carriers, patients with CNS onset reached renal failure markedly faster than those with infantile NS onset (median 0.5 months vs. 9.1 months; p<0.001).²
In a North American PNRC cohort of 11 centers (n=36), NPHS1 compound heterozygous variants — including splice site variants such as c.2335-1 G>A — were associated with higher proteinuria levels (112.4±135.6 vs. 53.9±57.3 g/day) and more severe proteinuria compared to patients with a single variant.¹ (Note: These figures are based on a small cohort and should be interpreted with caution.)
2-2. WT1: More Prevalent in Infantile NS; Variant Location Determines Prognosis
WT1 variants are found more frequently in the infantile NS group.¹ In the Japanese cohort, all CNS patients with WT1 variants had a median age of renal failure onset of 1.5 months, compared to 15.0 months in infantile NS patients — a statistically significant difference (p=0.006).²
Variant location also matters. Patients with variants located at the DNA-binding site in exon 8 or 9 had worse renal outcomes than those with variants at other positions; variants affecting the Cys2-His2 zinc finger structure were also associated with a severe phenotype.²
According to the ERKNet-ESPN consensus, pretransplant nephrectomy should be considered in patients with a dominant pathogenic WT1 variant.³ This illustrates how genetic testing results can directly influence surgical decision-making.
2-3. Age of Onset Is Itself a Prognostic Factor
Consistent differences in outcome between CNS (0–3 months) and infantile NS (3–12 months) are observed even within the same gene. In the Japanese multicenter study, among patients with non-NPHS1 variants, the median age at renal failure was 1.0 month in CNS patients versus 15.0 months in infantile NS patients (p<0.001).² This suggests that earlier onset is likely associated with more severe genotypes (e.g., truncating variants, severe missense variants).

─────────────────────────────
- Digenic and Triallelic Inheritance: Limitations of Single-Gene Testing
One pattern warrants special attention. In digenic inheritance — where pathogenic variants are present simultaneously in two genes such as NPHS1/WT1 or NPHS1/NPHS2 — patients are strongly associated with the need for transplantation regardless of age of onset, compared to those with a single-gene variant alone. This was reported in the North American PNRC cohort.¹
This has important clinical implications. For example, even if a single NPHS2 test returns negative, a severe phenotype may still emerge if a concurrent NPHS1 variant is present. “Negative for a single gene” does not mean “not genetic,” and this data supports the need for comprehensive panels or Whole Exome Sequencing (WES).
─────────────────────────────
- How Genetic Test Results Change Clinical Decision-Making
4-1. Preventing Unnecessary Immunosuppressive Exposure
Among 45 patients with confirmed variants in the four core genes (NPHS1, NPHS2, WT1, LAMB2) presenting with NS within the first year of life, only one showed a sustained response to steroids.¹ This data demonstrates that a response to immunosuppression is unlikely when a genetic NS is confirmed, and underscores the need for early genetic diagnosis to prevent the adverse effects associated with prolonged steroid exposure.
4-2. Direct Role in Transplant Strategy
Per the ERKNet-ESPN consensus recommendations, nephrectomy before kidney transplantation should be considered in patients with a dominant pathogenic WT1 variant³ — owing to the risk of Wilms tumor associated with WT1 variants. Conversely, patients with biallelic truncating NPHS1 variants may develop anti-nephrin antibodies following transplantation, requiring a different post-transplant immune monitoring strategy. Proceeding with transplantation without knowing the genotype risks missing these considerations.
4-3. Basis for Family Genetic Counseling
Both NPHS1 and NPHS2 follow an autosomal recessive inheritance pattern. Accordingly, in families with a confirmed proband, siblings carry a 25% risk of developing the disease, opening the door to genetic counseling for carrier parents and the possibility of prenatal diagnosis. These family-level interventions are not possible without a genetic diagnosis.
─────────────────────────────
- Current Limitations
Important knowledge gaps remain in this field. First, a substantial proportion of CNS and infantile NS cases still lack an identified monogenic cause, suggesting the possibility of undiscovered causative genes. Second, interpreting Variants of Uncertain Significance (VUS) remains a significant challenge. VUS have been reported in genes such as NPHS1 and CRB2, and establishing the pathogenicity of these variants can be difficult.⁵ Third, the limited availability of variant databases from non-European cohorts — including Asian and African populations — constrains the interpretation of population-specific variants.
─────────────────────────────
For patients with suspected congenital or infantile nephrotic syndrome, identifying the causative gene is the first clinical decision. 3billion offers Whole Exome Sequencing (WES) and Whole Genome Sequencing (WGS), together with automated reanalysis technology. Please use the button below for more information.
─────────────────────────────
[References]
- Islam MS et al. “Congenital and infantile nephrotic syndrome: genotype-phenotype associations.” Pediatric Research. 2025 – Rationale for Use: Gene distribution by onset age, correlation between compound heterozygous NPHS1 variants and proteinuria levels, and the association between digenic inheritance and transplantation outcomes.
- Inoki Y et al. “Differences in kidney prognosis between congenital and infantile nephrotic syndrome.” Pediatric Nephrology. 2025 — Rationale for Use: Median time to reach renal failure in CNS vs. infantile NS (NPHS1: 31.0 vs. — months / non-NPHS1: 1.0 vs. 15.0 months), renal prognosis based on WT1 variant locations, and the interaction between onset timing and non-NPHS1 genetic variants.
- Boyer O et al. “Management of congenital nephrotic syndrome: consensus recommendations of the ERKNet-ESPN Working Group.” Nature Reviews Nephrology. 2021;17(4):277-289. — Rationale for Use: Recommendation to consider pre-transplant nephrectomy in patients with WT1 dominant pathogenic variants.
- Lee JX et al. “NPHS Mutations in Pediatric Patients with Congenital and Steroid-Resistant Nephrotic Syndrome.” International Journal of Molecular Sciences. 2024 Nov 15;25(22). — Rationale for Use: Meta-analysis of NPHS1 and NPHS2 variant prevalence, risk of ESRF in European patients with NPHS2 variants (OR=7.97), evidence that four genes account for two-thirds of NS cases within the first year of life, and the finding that only 1 out of 45 patients with confirmed genetic variants responded to steroids.
- Zafar F et al. “Heterozygous variants of uncertain significance in NPHS1 and CRB2 in a newborn with congenital nephrotic syndrome of the Finnish type and multiple fetal anomalies: a case report.” AME Case Reports. 2025 — Rationale for Use: Clinical cases highlighting the limitations in interpreting Variants of Uncertain Significance (VUS).
- Rong L et al. “Genetic Variations and Clinical Features of NPHS1-Related Nephrotic Syndrome in Chinese Children: A Multicenter, Retrospective Study.” Frontiers in Medicine. 2021;8:771227. — Rationale for Use: Evidence showing that compound heterozygous NPHS1 variants are the predominant variant type in the Chinese population, within the context of ethnic differences in variant distribution.
Get exclusive rare disease updates
from 3billion.

Soo-jung Baek
As a marketer, I strive to empower the rare disease community by sharing meaningful insights backed by our company’s expertise.