Episode 11: [Rubinstein-Taybi Syndrome] Broad Thumbs, Developmental Delay, and Everything About “Transcriptional Disorders”
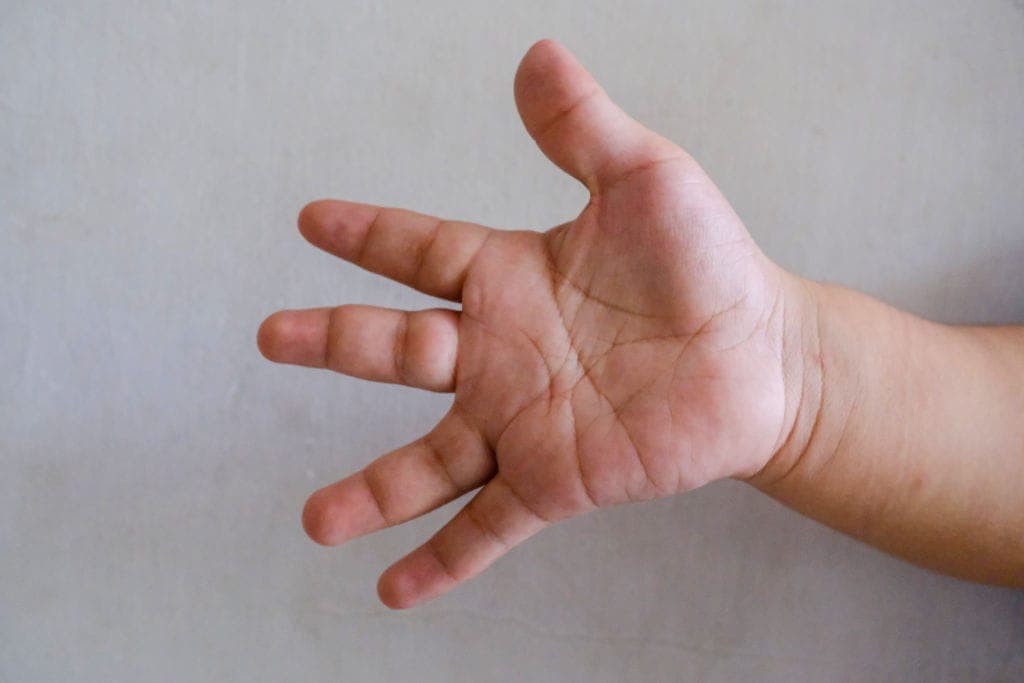
“My child has unusually broad and short thumbs and halluces compared with peers. His facial features look somewhat distinctive, and his development is slow. Why do such varied symptoms appear across the whole body rather than in just one organ?”
Many rare genetic disorders arise from a dysfunction of a single protein or a single biological pathway. Rubinstein-Taybi syndrome (RSTS), however, belongs to the group of chromatinopathies—disorders in which the abnormality lies not in a single protein but in the chromatin remodeling and transcriptional regulation system that controls the entire cellular gene-expression program. Because of this, RSTS and other chromatinopathies typically present with clinical manifestations that involve multiple organ systems simultaneously.
In this article, we take a deep look at how the RSTS-causing genes CREBBP and EP300 function in the body, and at the latest clinical trend in which the location of a variant within the same gene can lead to an entirely different diagnosis.
🔍 Hallmark Clinical Features of Rubinstein-Taybi Syndrome
Because RSTS results from a breakdown of the transcriptional regulatory system, it affects the development of virtually every organ from the embryonic stage. It should therefore be understood not as a single-organ disorder but as a systemic, multisystem developmental disorder.
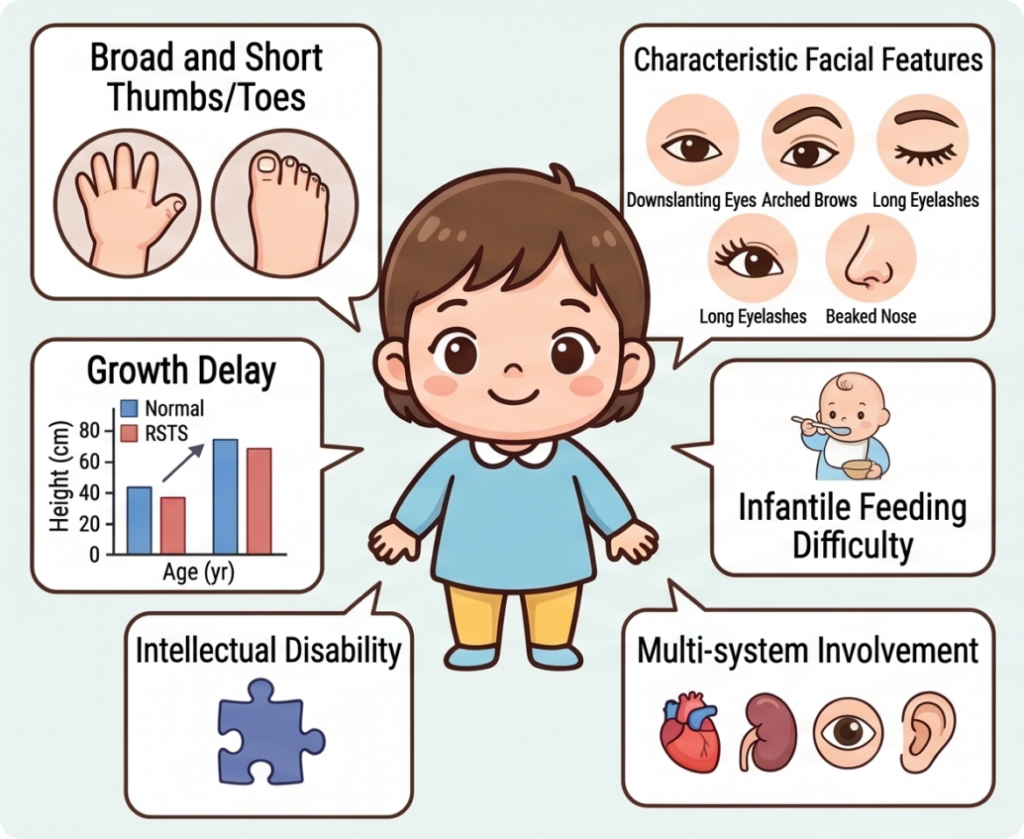
1. Physical and Morphological Features
- Broad thumbs and halluces: The most strongly suggestive physical hallmark of RSTS. Patients present with broad, short thumbs and great toes, often accompanied by a variety of skeletal anomalies.
- Characteristic facial features: Downslanting palpebral fissures, highly arched eyebrows, long and thick eyelashes, a prominent or beaked nose, a highly arched palate, and mild micrognathia are among the distinctive craniofacial and oral findings.
- Growth retardation and short stature: After infancy, growth velocity typically slows and short stature relative to peers is common; growth delay may persist over time.
2. Neurological and Systemic Features
- Developmental delay and intellectual disability: Motor and language milestones are delayed from infancy, and intellectual disability of varying severity is frequently observed.
- Feeding difficulties and language delay in infancy: Feeding difficulties and poor weight gain are common during infancy, and language development is often delayed.
- Multisystem organ involvement: Congenital heart defects, renal and urinary tract anomalies, ophthalmologic conditions (strabismus, refractive errors, cataracts, etc.), and hearing impairment can all occur across multiple organ systems.
- Skin abnormalities including keloids: Keloid formation—where scars from trauma or surgery grow abnormally large—is a relatively characteristic finding.
So why do RSTS1 and RSTS2 differ in phenotype despite sharing the same molecular mechanism?
Current evidence suggests that RSTS2, caused by EP300 variants, tends on average to present with a somewhat milder phenotype than RSTS1, which is caused by CREBBP variants. In RSTS2 patients, intellectual disability is generally less severe, and features such as broad thumbs/halluces and the characteristic facial gestalt are often less pronounced. Growth impairment also tends to be milder compared with RSTS1.
These differences are thought to arise from a combination of factors—tissue-specific functional differences between CREBBP and EP300, differences in the transcription factors each interacts with, and residual protein function—though the molecular basis has not yet been fully clarified. Because the phenotypes of the two conditions also overlap substantially, it is difficult to reliably distinguish RSTS1 from RSTS2 on clinical grounds alone.
The Normal Function of CREBBP and EP300: Master Platforms for Transcriptional Activation
The molecular etiology of Rubinstein-Taybi syndrome lies in pathogenic variants in two genes: CREBBP (RSTS type 1, ~55–75%) and EP300 (RSTS type 2, ~8–11%). The two genes are paralogs that arose through gene duplication during evolution and share a remarkably high 86% amino acid sequence identity within their core histone acetyltransferase (KAT) domain.
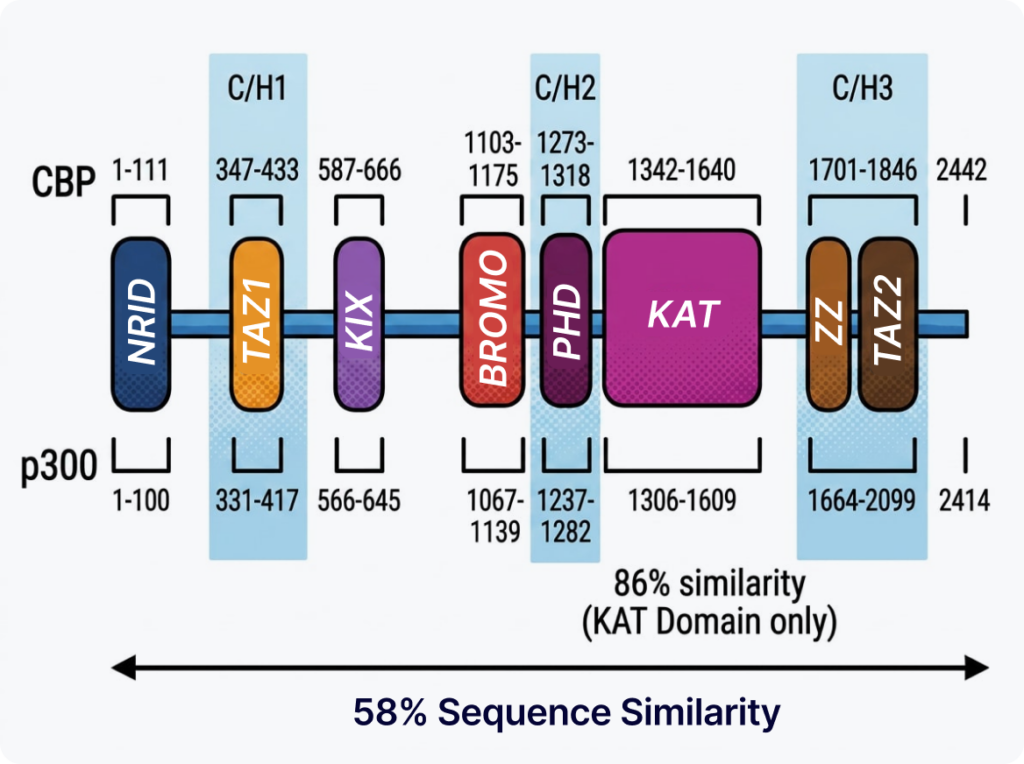
The proteins they encode—CBP and p300—act as master co-activator platforms that drive gene transcription within the cell, performing three major molecular functions.
1. H3K27 Acetylation: Enhancer Activation
CBP and p300 acetylate lysine 27 of histone H3 (H3K27ac), loosening chromatin structure and keeping enhancer regions in an active state. This creates the conditions in which the developmental genes required for embryonic development, nervous system formation, and organogenesis can be expressed at the right time. H3K27ac is a canonical epigenetic marker of active enhancers.
2. Scaffold for Transcription Complex Assembly
CBP and p300 are not enzymes alone—they also act as scaffolds that link multiple proteins together. By connecting various transcription factors, the Mediator complex, and RNA polymerase II into a single assembly, they help ensure that the transcription complex forms stably. This bridging function allows transcription to be initiated efficiently and regulated with precision.
3. Non-histone Protein Acetylation
The acetylation activity of CBP and p300 is not restricted to histones. These enzymes also acetylate a range of transcription factors and regulatory proteins—including p53 and RUNX2—modulating their stability, activity, and DNA-binding capacity. Through this, essential biological processes such as cell proliferation, differentiation, skeletal formation, neural development, and cellular stress responses are maintained.
The Pathophysiology of Rubinstein-Taybi Syndrome: A Cascading Collapse of Three Functions
So what happens inside the cell when pathogenic variants in CREBBP or EP300 prevent these functions from being carried out properly? The pathophysiology of RSTS begins with the sequential collapse of the three functions described above.
1. Reduced H3K27 Acetylation: Impaired Enhancer Function
The core pathophysiology of RSTS is haploinsufficiency. When loss-of-function variants occur in CREBBP or EP300, the amount of functional CBP/p300 protein decreases, and histone acetyltransferase (HAT) activity is reduced.
In normal cells, acetylation by CBP/p300 and deacetylation by HDACs (histone deacetylases) are balanced so that active enhancers are maintained. In RSTS, however, H3K27 acetylation cannot be carried out adequately, H3K27ac levels decline, chromatin gradually condenses (chromatin compaction), and chromatin accessibility is reduced. As a result, enhancers that would normally remain active can no longer sustain their function, and expression of developmentally important genes is broadly diminished.
2. Impaired Scaffold Function: Defective Transcription Complex Assembly
CBP and p300 are not simple enzymes—they act as transcriptional co-activators and scaffolds that connect multiple proteins. By binding a diverse set of transcription factors such as CREB, p53, STAT, β-catenin, and HIF-1α, they recruit the Mediator complex and RNA polymerase II and help the transcription complex assemble stably.
In RSTS, the shortage of functional CBP/p300 undermines this scaffold role as well. Even when transcription factors bind their target genes, the Mediator complex and RNA polymerase II are not recruited efficiently, and assembly of the transcription complex becomes unstable. Ultimately, the efficiency of transcription initiation drops, and expression of the many genes required for development and differentiation declines.
3. Reduced Non-histone Protein Acetylation: Diminished Transcription Factor Activity
CBP and p300 acetylate not only histones but also non-histone proteins such as p53, RUNX2, STAT, and β-catenin, regulating their stability, activity, and DNA-binding capacity. These processes are essential for normal cell proliferation and differentiation, skeletal formation, neural development, and other biological functions.
In RSTS, reduced CBP/p300 acetyltransferase activity leads to a corresponding reduction in the acetylation of these non-histone proteins. As a consequence, the activity of numerous transcription factors and regulatory proteins declines, and signaling pathways fail to operate normally. Combined with impaired enhancer function and defective transcription complex assembly, this further deepens the downregulation of developmental genes and is thought to contribute to the neurodevelopmental abnormalities, skeletal anomalies, growth impairment, and multiorgan phenotypes seen in RSTS.
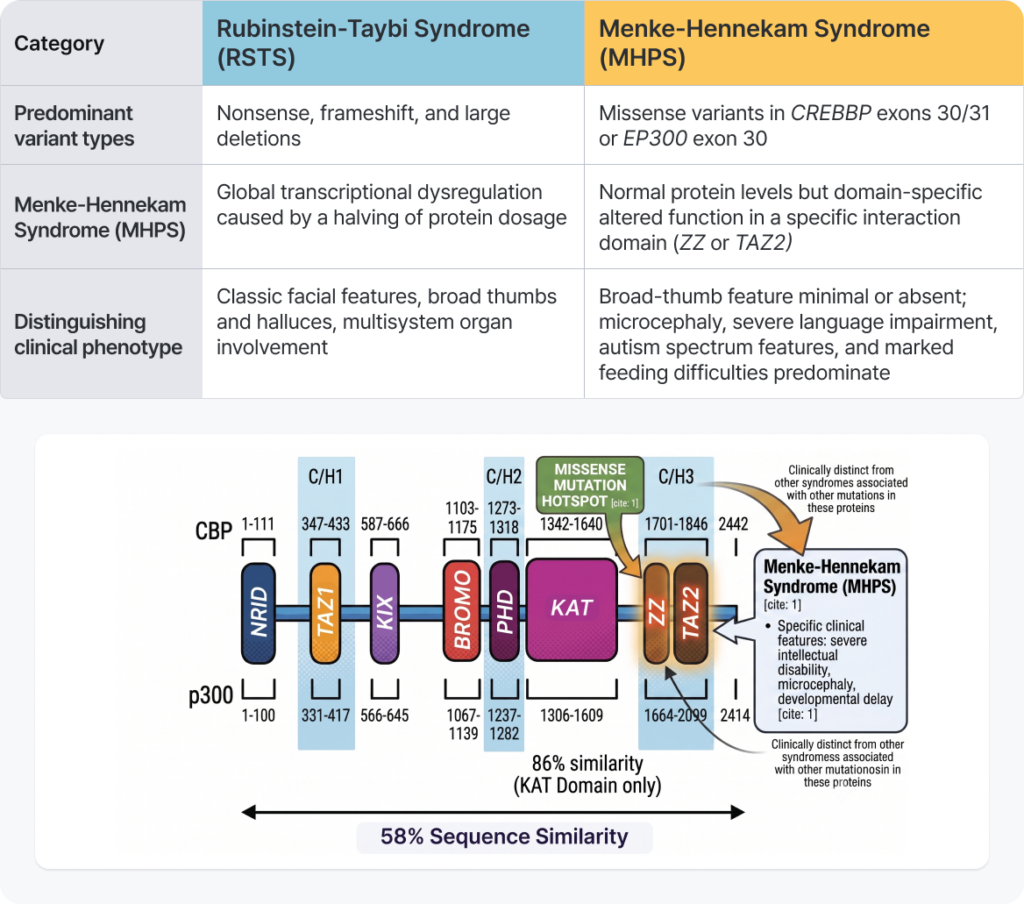
The Reversal Driven by Variant Type: Differentiating Menke-Hennekam Syndrome
The most important point to recognize in variant analysis is that the nature of the variant (variant type) and its location within the gene can drive dramatically divergent phenotypes.
Gene Variant Mechanism Comparison Guide
How Is a Clinical Diagnosis Made?
Although next-generation sequencing (NGS) now makes it possible to identify CREBBP and EP300 variants directly, clinical diagnostic criteria remain highly important.
Clinical assessment is particularly valuable in situations such as:
- When a variant of uncertain significance (VUS) is identified and its pathogenicity must be judged
- When Rubinstein-Taybi syndrome (RSTS) is strongly suspected on clinical grounds but molecular diagnosis has not been achieved
- When genetic testing is not readily accessible
To address these needs, a clinical scoring system based on cardinal features and supportive features has been proposed for RSTS. Each clinical finding is assigned points, and the total score classifies the likelihood of RSTS into four tiers:
- 0–4 points: Unlikely RSTS
- 5–7 points: Possibly RSTS
- 8–11 points + cardinal feature: Likely RSTS
- ≥12 points + cardinal feature: Definitely RSTS
This scoring system has demonstrated strong diagnostic performance in validation studies. When applied to 100 patients with molecularly confirmed RSTS, all patients scored ≥5, yielding a sensitivity of 100%, while among 45 patients with Menke-Hennekam syndrome—which involves the same CREBBP/EP300 genes but is a distinct disorder—not a single patient was classified as Likely or Definitely RSTS.
The scoring system therefore serves as a highly useful tool for both effectively identifying RSTS patients and distinguishing RSTS from closely related conditions such as Menke-Hennekam syndrome.
The Compass for Precision Variant Interpretation: Efficient VUS Analysis with GEBRA
Rubinstein-Taybi syndrome is not merely a single-organ dysfunction—it is a complex genetic disorder in which the entire chromatin remodeling system, the master switch of gene transcription across every cell in the body, is affected. Because the direction of disease is completely reshaped by fine-grained variant location within specific domains and by variant type (LoF vs. missense), the outcome of diagnosis hinges on the ability to interpret variant sequence data precisely and from multiple angles.
At the heart of overcoming the limits of conventional clinical genomic screening filters—which struggle to detect the subtle functional impact of fine-domain variants and to resolve VUS—and of maximizing the accuracy of genomic interpretation is the GEBRA precision genomic analysis solution.
GEBRA goes beyond simple variant filtering: it is a clinical variant interpretation platform that analyzes disease mechanisms and variant types together on a per-gene basis. Users can intuitively review variant characteristics such as loss-of-function, missense, and splice variants, judge pathogenicity more quickly, and efficiently identify the diseases most likely associated with each variant. This raises the efficiency of VUS interpretation and supports more accurate clinical decision-making.
If you are facing challenges in clinical reporting due to ambiguous variants of uncertain origin, or if you would like to dramatically improve the efficiency of raw sequencing data analysis in your laboratory, explore the advanced precision genomic analysis solution from GEBRA today.
Frequently Asked Questions (FAQ)
Q1. How do CREBBP-related (type 1) and EP300-related (type 2) RSTS differ in phenotypic severity?
A. Because the two conditions share the same molecular pathway, their clinical features overlap substantially. However, large cohort data indicate that type 1 (CREBBP) patients show a more classic facial gestalt, more frequent skeletal and skin abnormalities, and relatively greater severity of cognitive impairment. Type 2 (EP300) patients, by contrast, tend to have milder external morphological features and, on average, statistically better cognitive function than type 1.
Q2. Even after molecular diagnosis (NGS), why do we still need a clinical scoring system?
A. Because the majority of variants identified in genomic data pass through the variant-of-uncertain-significance (VUS) stage. When the pathogenicity of a detected variant is unclear, a high quantified clinical scoring result can serve as strong phenotypic evidence in ACMG guideline-based interpretation. In addition, when a patient shows a classic phenotype but NGS is negative due to intronic or non-coding variants, or when differentiating RSTS from Menke-Hennekam syndrome, the scoring system becomes a critical benchmark that complements the accuracy of the analytical pipeline.
Q3. How can we visually explain the transcriptional-activation platform mechanism of CBP and p300?
A. If we imagine chromatin as a giant closed-stack library system, CBP and p300 are not merely transcription factors that read gene code. They are, in essence, the platforms that “unlock and open the shelves (histone acetylation) and dock the many transcriptional enzyme complexes required to copy the gene at the correct positions (scaffold function).” When haploinsufficiency dismantles this platform, the entire library remains locked in a silenced state, and the physical initiation of transcription for downstream developmental genes is effectively blocked.
References
Yazıcı E, et al. The complex network of p300/CBP regulation: Interactions, post-translational modifications, and implications in human disease. Journal of Biological Chemistry. 2025;301(11):110715. doi:10.1016/j.jbc.2025.110715.
Milani D, Manfredi M, Ajmone PF, et al. Rubinstein–Taybi syndrome: clinical features, genetic basis, diagnosis, and management. Journal of Medical Genetics. 2024;61(6):503–519. doi:10.1136/jmg-2023-109438.
Valor LM, Viosca J, Lopez-Atalaya JP, Barco A. Lysine acetyltransferases CBP and p300 as therapeutic targets in cognitive and neurodegenerative disorders. Neurotherapeutics. 2013;10(4):568–588. doi:10.1007/s13311-013-0204-7.
Get exclusive rare disease updates
from 3billion.

3billion Inc.
3billion is dedicated to creating a world where patients with rare diseases are not neglected in diagnosis and treatment.