“El síndrome de Usher está más cerca de lo que crees” —Una conversación con el Dr. Juan Carlos Zenteno Ruíz

📍Puntos clave
1) El diagnóstico temprano del síndrome de Usher está directamente relacionado con los resultados del tratamiento. Un diagnóstico preciso determina si puede iniciarse la rehabilitación y marca el punto de partida para el asesoramiento genético de toda la familia.
2) Las pruebas genéticas integrales son esenciales. Dada la variabilidad fenotípica y la existencia de condiciones similares, el estudio genético —incluyendo el WES— es la herramienta clave para confirmar el diagnóstico de síndrome de Usher.
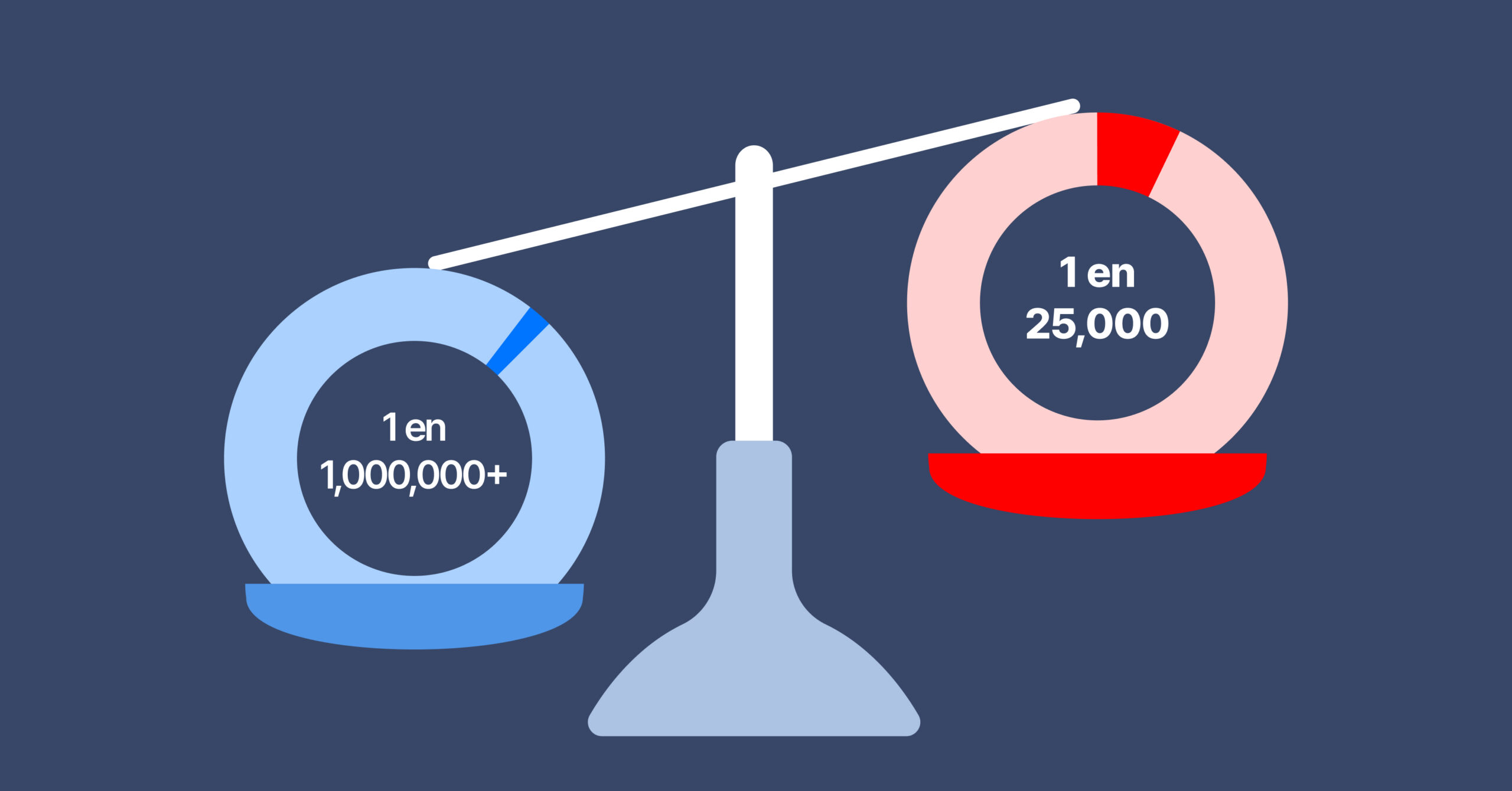
El síndrome de Usher está clasificado como una enfermedad rara, pero está lejos de serlo en comparación con la mayoría de las condiciones dentro de esta categoría. Mientras que la mayoría de las enfermedades raras afectan aproximadamente a 1 de cada 1,000,000 de personas, la prevalencia global del síndrome de Usher se estima en aproximadamente 1 de cada 25,000. Esto refleja el papel significativo que esta condición tiene en la práctica clínica.
En esta publicación, conversamos con el Dr. Juan Carlos Zenteno —especialista en genética, profesor de la Facultad de Medicina de la UNAM, director de la Unidad de Diagnóstico de Enfermedades Raras (UDER) y autor de más de 230 publicaciones internacionales— para explorar la realidad clínica del síndrome de Usher. A partir de su experiencia en el contexto clínico mexicano, analizamos cómo sospechar, diagnosticar y abordar el estudio genético de esta condición.
Para ver el perfil completo del Dr. Zenteno, consulte la parte inferior de esta página.
Sección 1 — ¿Qué es el síndrome de Usher?
El síndrome de Usher es un trastorno genético autosómico recesivo que provoca simultáneamente distrofia retiniana y pérdida auditiva neurosensorial. En algunos subtipos, también puede presentarse disfunción vestibular (alteración del equilibrio).
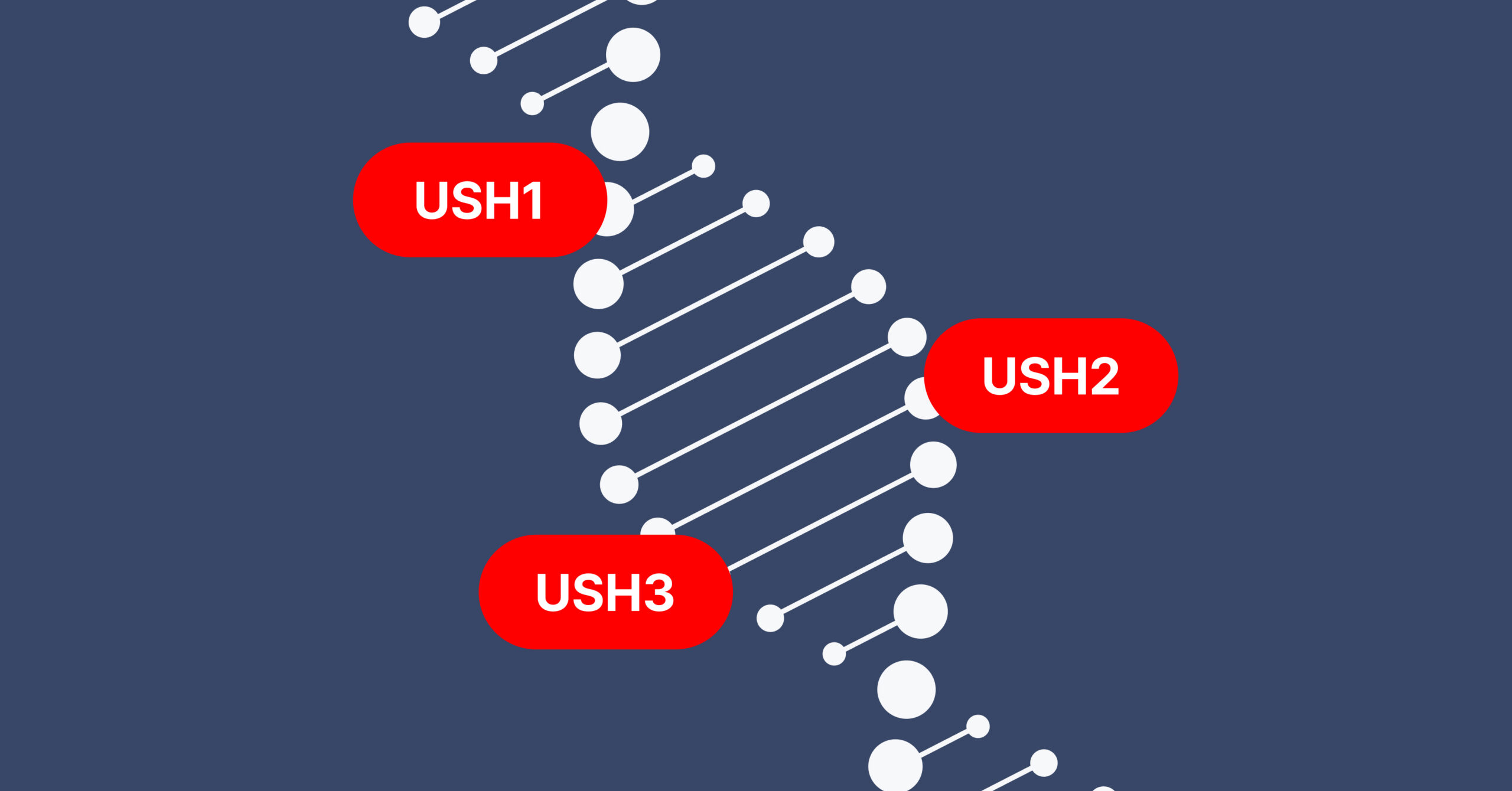
Subtipos clínicos
El síndrome de Usher se clasifica en tres subtipos principales según su gravedad y el momento de aparición:
- Tipo 1 (USH1): Sordera profunda congénita, disfunción vestibular desde el nacimiento y aparición de retinosis pigmentaria antes de la pubertad. Es el subtipo más severo, con una penetrancia del 99–100%.
- Tipo 2 (USH2): Pérdida auditiva congénita de moderada a severa con función vestibular normal. La retinosis pigmentaria suele desarrollarse en la adolescencia. Es el subtipo más común (aproximadamente 60% de los casos), con variantes en USH2A que representan el 80–90% de los casos de USH2.
- Tipo 3 (USH3): Pérdida auditiva progresiva y distrofia retiniana, con afectación vestibular variable. Se reporta con mayor frecuencia en ciertas regiones como Finlandia.
Entonces, ¿cómo se presenta realmente el síndrome de Usher en la práctica clínica? Le preguntamos directamente al Dr. Juan Carlos Zenteno Ruíz.
Sección 2 — Entrevista: Dr. Juan Carlos Zenteno Ruíz en la primera línea clínica
P. ¿Con qué frecuencia encuentra casos sospechosos o confirmados de síndrome de Usher en la práctica clínica? ¿Existen características que permitan sospecharlo tempranamente?
En mi experiencia el síndrome de Usher es uno de los diagnósticos más frecuentes en pacientes con distrofias de retina. De hecho, en nuestro servicio es la causa más frecuente de retinosis pigmentaria de tipo sindrómica, por arriba de enfermedades como el síndrome de Bardet-Biedl o el síndrome de Alstrom.
Este síndrome es la causa más común de la asociación deficiencia-visual, por lo que debe sospecharse en todo paciente con distrofia de retina e hipoacusia de inicio temprano.
P. ¿Por qué es importante un diagnóstico temprano y preciso en pacientes con sospecha de síndrome de Usher?
El diagnóstico temprano permite definir de manera precisa el diagnóstico y excluir otras enfermedades genéticas que cursan con la asociación de distrofia de retina e hipoacusia. El reconocimiento temprano del síndrome permite intervenciones de rehabilitación visual y ayudas auditivas en estos pacientes, así como un asesoramiento genético preciso a la familia.
P. ¿Cuáles son los principales desafíos en el diagnóstico del síndrome de Usher?
El principal desafío es la variabilidad fenotípica intra e interfamiliar, lo que puede llevar a retrasos en el diagnóstico clínico. Además, la combinación de distrofia de retina-hipoacusia ocurre en otras enfermedades monogénicas por lo que debe de realizarse una valoración clínica completa seguida de pruebas genéticas apropiadas.
Un análisis más profundo: ¿Qué impulsa el retraso diagnóstico en el síndrome de Usher?
La variabilidad fenotípica está ampliamente respaldada en la literatura. La heterogeneidad genética es un factor clave: un mismo gen puede generar diferentes manifestaciones clínicas, e incluso hermanos con la misma mutación pueden presentar cuadros distintos.
En el caso del USH2, la pérdida auditiva puede detectarse desde la infancia, mientras que la retinosis pigmentaria puede aparecer hasta la adolescencia. Esta diferencia temporal puede hacer que se pierda la oportunidad diagnóstica si no existe una sospecha clínica adecuada.
Diagnóstico diferencial incluye:
- Síndrome de Heimler: Hipoacusia profunda, distrofia de retina y anomalía dentales.
- Síndrome de Bardet-Biedl: distrofia retiniana, obesidad, polidactilia y discapacidad intelectual. La pérdida auditiva es menos prominente.
- Síndrome de Alström: distrofia retiniana y pérdida auditiva con compromiso multisistémico (obesidad, diabetes, cardiomiopatía).
- Síndrome PHARC: condición muy rara con polineuropatía, pérdida auditiva, ataxia cerebelosa, retinosis pigmentaria y cataratas. (Harutyunyan et al., Orphanet J Rare Dis, 2024.DOI)
- Coincidencia de retinosis pigmentaria no sindrómica con pérdida auditiva hereditaria independiente.
Esta superposición clínica demuestra que la evaluación clínica por sí sola no es suficiente. La combinación de variabilidad fenotípica y diagnósticos similares refuerza que las pruebas genéticas no son opcionales, sino esenciales para confirmar el diagnóstico.
P. ¿En qué momento del proceso diagnóstico deberían considerarse las pruebas genéticas en pacientes con sospecha de síndrome de Usher? ¿Recomendaría realizarlas incluso antes de que aparezcan síntomas visuales claros en pacientes con pérdida auditiva?
Inmediatamente después de la sospecha diagnóstica y de haber excluido causas no genéticas del fenotipo (infecciones, fármacos).
Lo recomendaría en casos en los que la pérdida auditiva sea temprana o que la historia familiar apoye una causa monogénica (padres consanguíneos o con endogamia, otros individuos afectados en la familia)
P. En su opinión, ¿cuáles son las ventajas de utilizar WES (secuenciación del exoma completo)?
A mi juicio, WES es la prueba ideal en pacientes con sospecha de síndrome de Usher o de hipoacusia de tipo monogénico ya que permite analizar la totalidad de los genes conocidos que se asocian a estos fenotipos. Además, los datos de WES pueden ser reanalizados periódicamente en los casos negativos ya que existe la posibilidad de la identificación de nuevos genes para el fenotipo o bien que ciertas variantes sean reclasificadas como causales de la enfermedad.
Si está considerando el WES, contáctenos a través del botón a continuación. Nuestros especialistas están listos para responder sus preguntas sobre el costo, el proceso y los materiales necesarios.
Perfil del coautor
Juan Carlos Zenteno Ruíz, MD, PhD
El Dr. Juan Carlos Zenteno Ruíz obtuvo su título de médico en la Universidad Juárez Autónoma de Tabasco, realizó su especialidad en Genética en el Hospital General de México y cuenta con una Maestría y un Doctorado en Genética por la Universidad Nacional Autónoma de México (UNAM).
Es Profesor de Tiempo Completo (Profesor C) en la Facultad de Medicina de la UNAM y miembro del Sistema Nacional de Investigadores (SNI) de México en el nivel más alto — Nivel 3 (2023–2032).
Actualmente se desempeña como Director de la Unidad de Diagnóstico de Enfermedades Raras (UDER) en la Facultad de Medicina de la UNAM, donde lidera investigaciones sobre las bases genéticas de enfermedades oculares raras y hereditarias en México.
Hasta la fecha, ha publicado más de 230 artículos internacionales sobre los fundamentos genéticos de enfermedades oculares raras y hereditarias, acumulando más de 5,000 citas. Además, imparte docencia tanto a nivel de licenciatura como de posgrado en la Facultad de Medicina de la UNAM.
Referencias
- Stéphanie Nguengang Wakap, Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database(2019) https://www.nature.com/articles/s41431-019-0508-0
- Mor Hanany, Comparison of Worldwide Disease Prevalence and Genetic Prevalence of Inherited Retinal Diseases and Variant Interpretation Considerations, (2024) https://perspectivesinmedicine.cshlp.org/content/14/2/a041277
Get exclusive rare disease updates
from 3billion.

Soo-jung Baek
As a marketer, I strive to empower the rare disease community by sharing meaningful insights backed by our company’s expertise.




