Diagnosing Myotonic Dystrophy: A Case of Cryptorchidism and Muscle Weakness

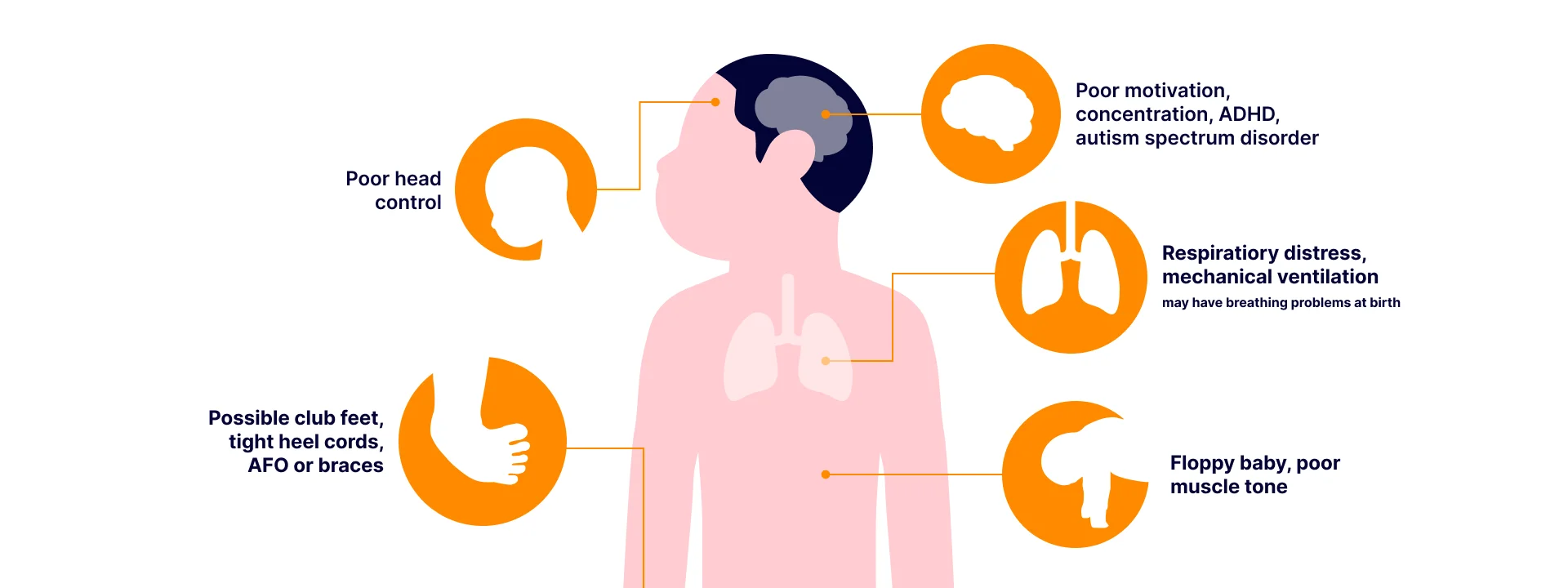
There was a male infant present with cryptorchidism and muscle weakness. Both parents of the child were unaffected, and there was no family history. The clinician suspected congenital myotonic dystrophy. The proband was an infant male present with cryptorchidism and muscle weakness. Both parents were unaffected and there was no family history. The clinician suspected congenital myotonic dystrophy.
Myotonic dystrophy is a disorder that affects the skeletal and smooth muscles of multiple systems. It is characterized by progressive wasting and weakening of muscles. Affected individuals often have prolonged muscle contraction, also they may suffer from slurred speech or a locked jaw. There are two types of myotonic dystrophy.

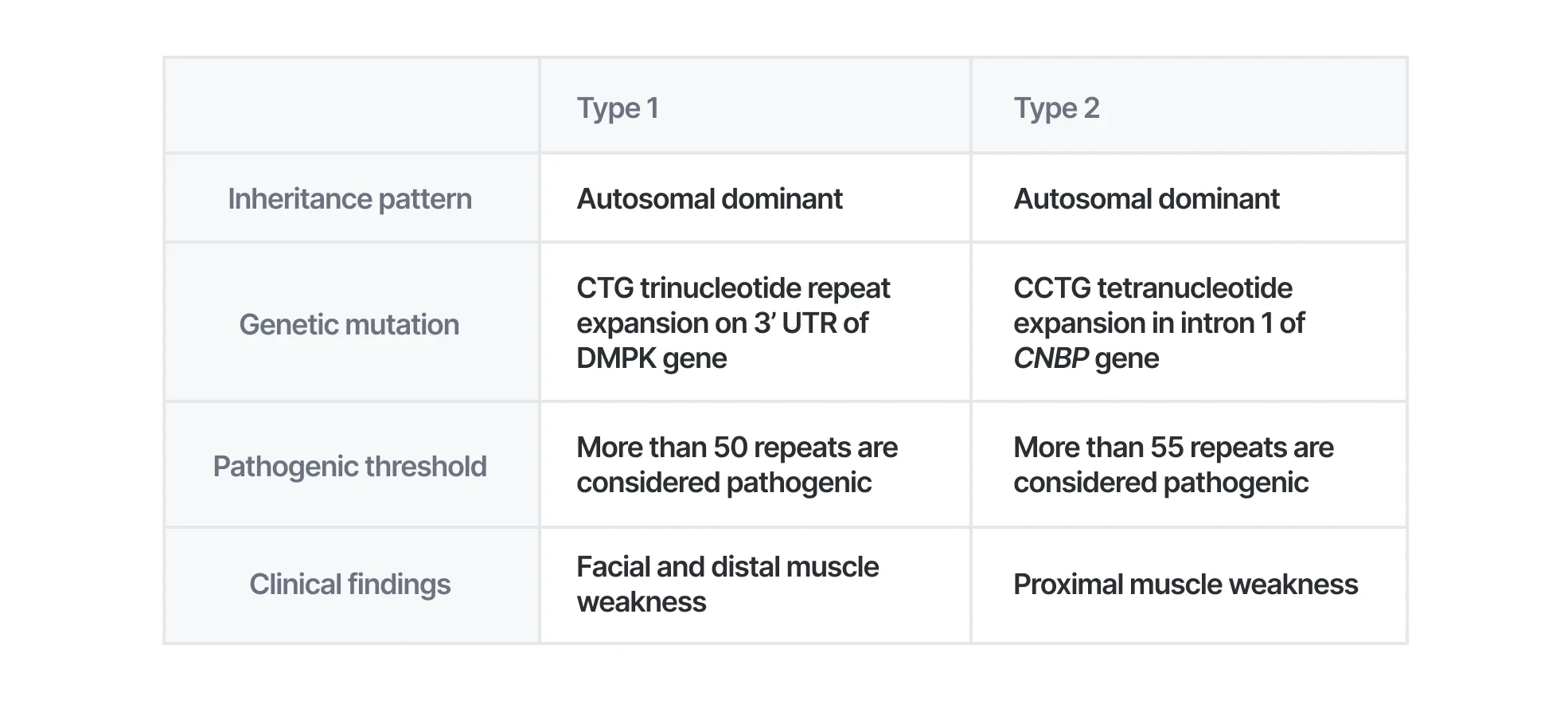
Myotonic dystrophy type 1 (DM1) is an autosomal dominant disorder caused by CTG repeat expansion in 3’ UTR of the DMPK gene. Normal alleles carry 5 to 34 CTG repeats while full-penetrance pathogenic alleles carry more than 50 CTG repeats. DM1 can be suspected if the patient has distal and facial muscle weakness, myotonia, and posterior subcapsular cataracts. In neonates, hypotonia, facial muscle weakness, generalized weakness, positional malformation, and respiratory insufficiency can be indicators for suspecting DM1.
Myotonic dystrophy type 2 (DM2) is also an autosomal dominant disorder that is caused by a heterozygous pathogenic expansion of a CCTG repeat with a complex repeat motif in CNBP. Repeats of CNBP are in the intron 1. Alleles with more than 55 CCTG repeats can be considered pathogenic. If an individual has muscle weakness, myotonia, posterior subcapsular cataract, cardiac conduction defect or progressive cardiomyopathy, insulin insensitivity, and hypogammaglobulinemia, DM2 should be suspected.
Both types of myotonic dystrophy are caused by repeat expansion in non-coding regions. This means WES can’t diagnose repeat expansion of DMPK and CNBP, since it only targets protein-coding regions.

Luckily, the patient and his family received whole genome sequencing. In this picture, you can see an increase in the depth of the repeated region in the 3′ UTR of the DMPK gene in the WGS result. However, in the WES result, an increase cannot be seen since the repeated region is not the target of capture. The WGS result was analyzed using Expansion Hunter (Illumina). The proband was estimated to have 123 CTG repeats, which is inherited from the mother who carries 90 CTG repeats.
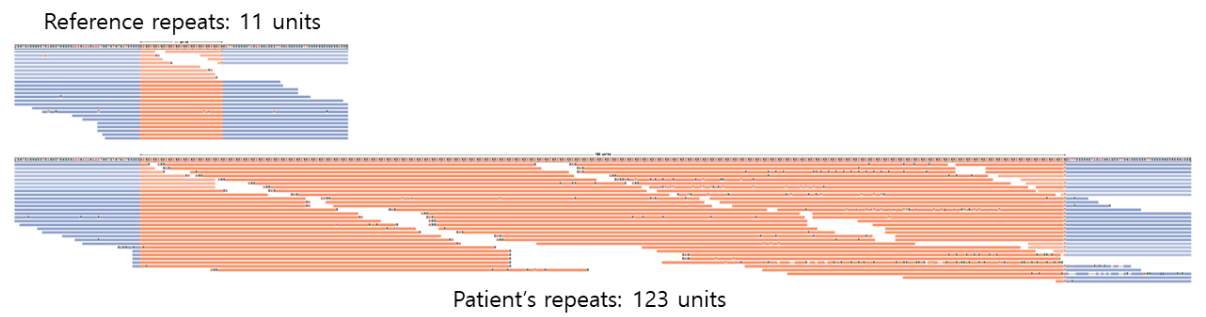
With phenotype suggesting myotonic dystrophy and the number of repeats in DMPK over the pathogenic threshold, the patient was diagnosed with myotonic dystrophy type 1.
There are some limitations for diagnosis of repeat expansion disorders. Since the number of repeats is estimated using expansion hunter, the exact number of repeats can’t be measured. Also it is impossible to estimate the number of repeats that are too long. For example, patients with congenital myotonic dystrophy 1 are reported to carry more than 1,000 CTG repeats. In this case, the estimated number of repeats were only 123, which is far below a thousand. These are the technical limitations that we are facing. For the cases of repeat expansion disorders, we recommend using our report as a strong suggestion for taking alternate clinical testing such as PCR fragment analysis or southern blotting.

Discover Our Advanced Whole Genome Sequencing!
Get the most detailed and accurate Genome Test available
If you are interested in this case, you might want to read:
1. https://3billion.io/blog/limitation-of-whole-genome-sequencing
2. https://3billion.io/blog/diagnostic-approach-for-huntington-disease
For more information about myotonic dystrophy, please read these:
1. Myotonic dystrophy type 1 (OMIM: 160900)
2. Myotonic dystrophy type 2 (OMIM: 602668)
Get exclusive rare disease updates
from 3billion.

Yongjun Song
Medical geneticist skilled in variant interpretation. Mainly interested in developing interpretation software including pipeline for rare disease diagnostics





