Diagnostic approach for Huntington disease
One day, 3billion received a blood sample from an elderly European woman suffering from ataxia, on which whole-exome sequencing (WES) was performed to determine its cause. Following examination, she was diagnosed with Huntington disease.
What is Huntington disease?
Huntington disease is an inherited, progressive, and neurodegenerative disorder characterized by multiple symptoms, including chorea, impaired coordination, difficulty speaking, cognitive decline, and other psychiatric symptoms. In the early stages of the disease, affected individuals often experience subtle symptoms, such as clumsiness, memory lapses, depression, and mood swings. As the disease progresses, movement disorders, such as unsteady gait and/or involuntary movement, appear and gradually aggravate.1 In most patients, symptoms usually appear in middle age but may appear earlier and more severely in some cases.
Understanding Huntington disease
To understand this disease, the following terms need to be comprehended: Huntingtin, tandem repeats, repeat expansion disorder, and genetic anticipation.
Huntington disease is caused by a mutation in Huntingtin. The Huntingtin contains a region where a series of glutamines is adjacently repeated. These glutamines are coded by repeated CAG codons called trinucleotide or tandem repeats.
Tandem repeats or microsatellites, are regions consisting of one or more nucleotides that are repeated directly adjacent to each other. The human genome carries numerous patterns of tandem repeats. These regions have high mutation rates, leading to high genetic diversity. Therefore, each of them differs from person to person2. Some tandem repeats are located within genes, and a few of those are associated with repeat expansion disorders.
Repeat expansion disorders refer to a group of disorders caused by an increased number of repeats within specific genes. The repeats may occur in the coding and non-coding regions of genes, such as UTRs and introns (Figure 1)3.
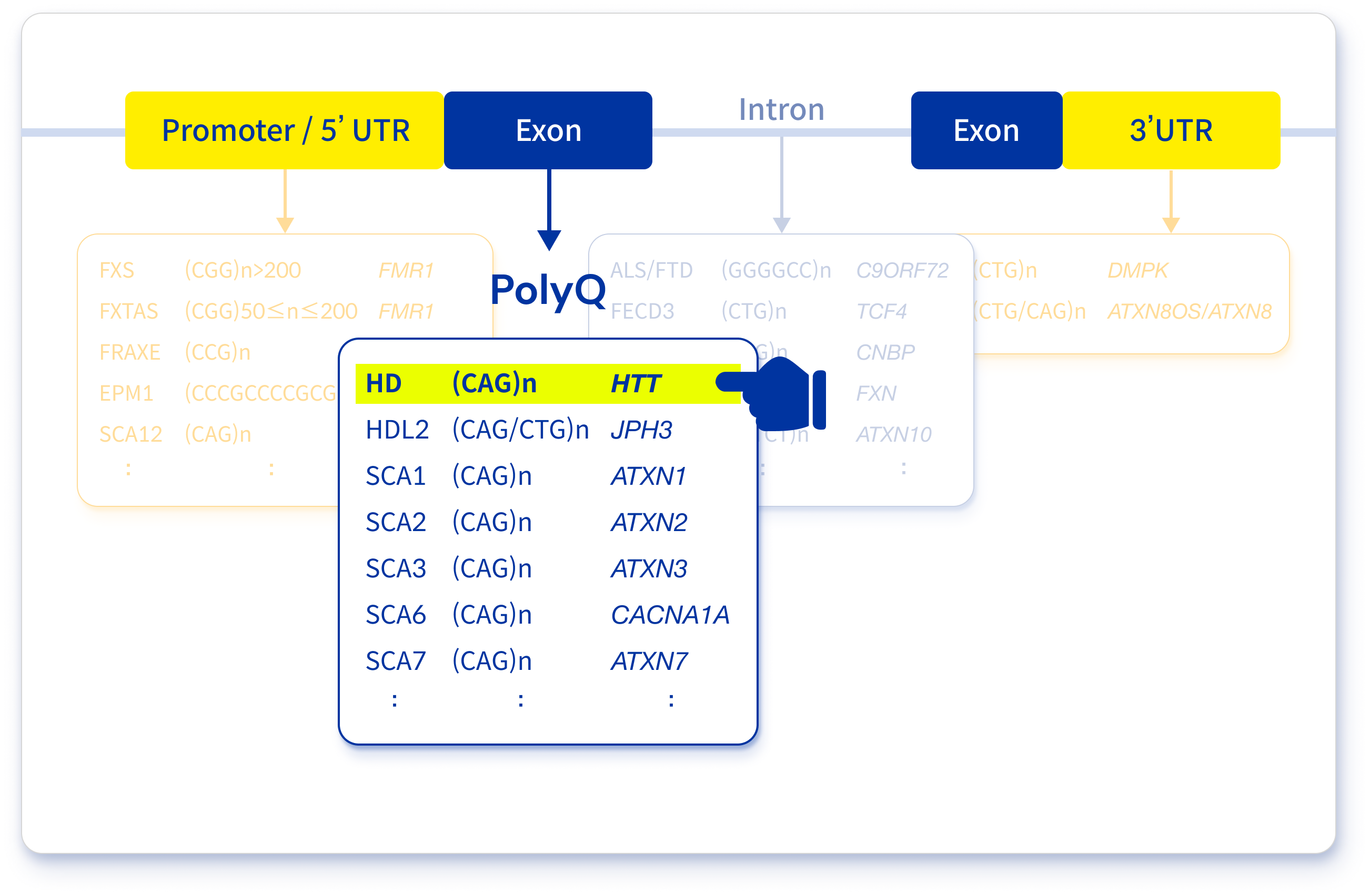
Figure 1. A list of repeat expansion disorders focused on polyglutamine diseases
Huntington disease is one of repeat expansion disorders. Specifically, this disease is one of the trinucleotide expansion disorders. A repeat of Huntington disease is located on exon 1 and codes the number of glutamines by multiple repeated CAG nucleotides. Generally, CAG repeats under 27 are considered normal. Furthermore, 27–35 repeats are considered normal but can be pathogenic in successive generations. Additionally, 36–39 repeats are considered pathogenic with reduced penetrance, meaning it may be pathogenic. From 40, it is considered pathogenic with full penetrance and a larger number of repeats, causing juvenile-onset Huntington disease (Table 1)4,5.
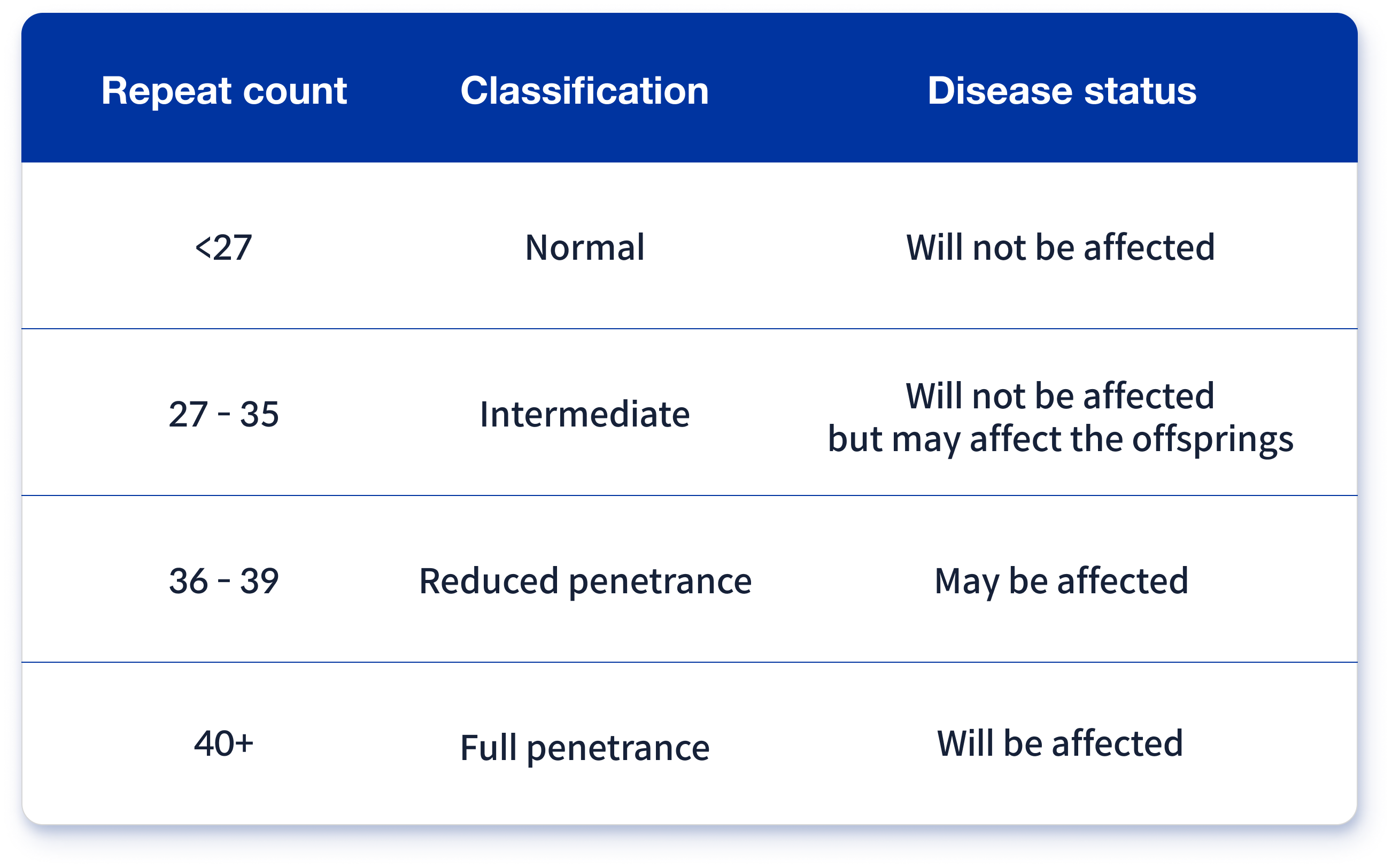
Table 1. Classification of Huntington disease based on repeat counts
Affected individuals produce Huntingtin with 36 or more glutamine repeats. This pathogenic protein is called mutated Huntingtin, which causes neurons of certain brain regions to decay, leading to signs and symptoms of Huntington disease.
The disadvantage of repeat expansion disorders, including Huntington disease, is that they become more severe as successive generations inherit them. The number of highly mutable repeats tends to increase as they are transmitted from parents to children; therefore, the consequences of the disorder are intensified in successive generations3. This characteristic of repeat expansion disorders is called genetic anticipation (Figure 2).
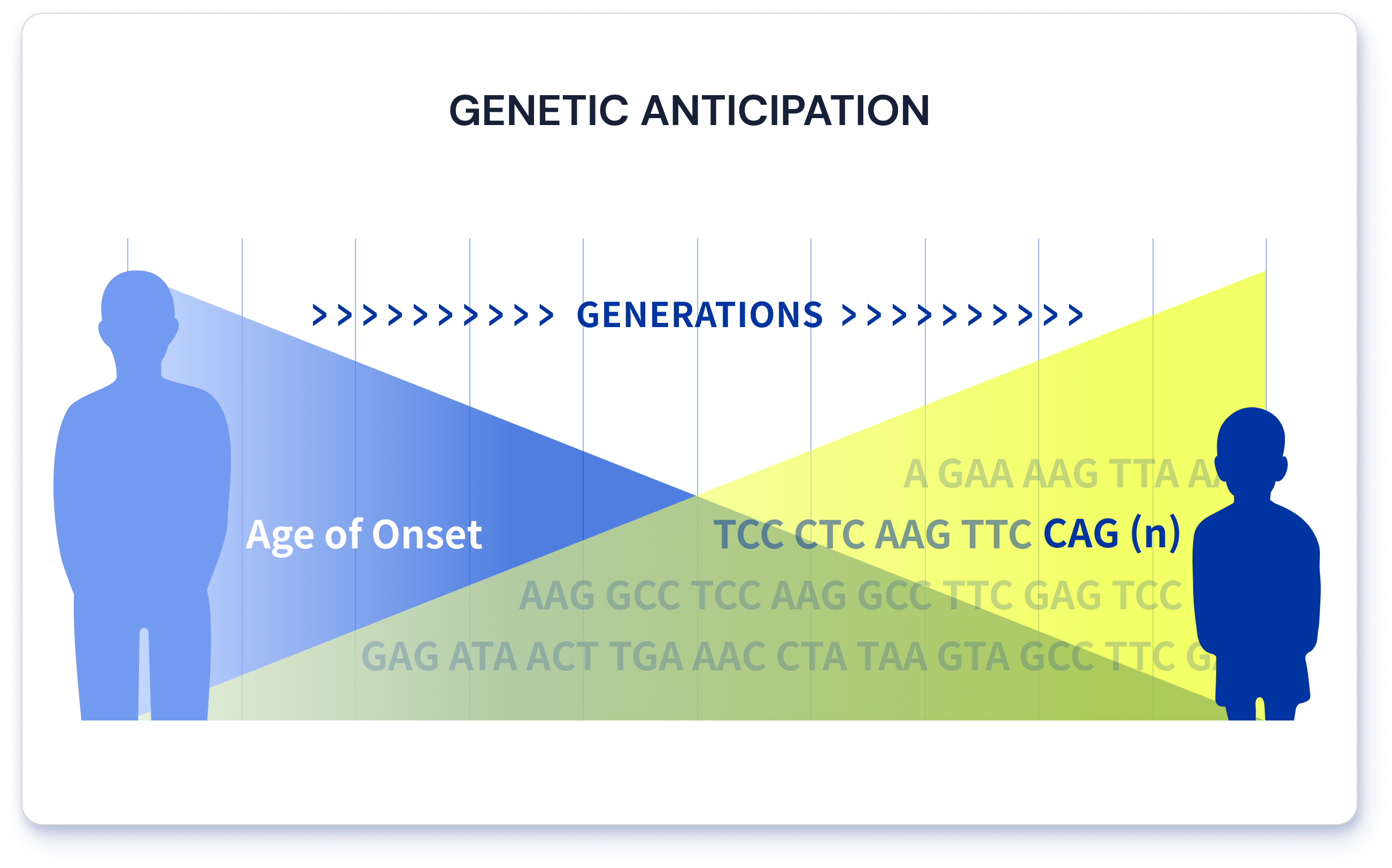
Figure 2. An illustration of genetic anticipation
How do we diagnose Huntington disease and other repeat expansion disorders?
To diagnose a patient with Huntington disease, two things are required, one is phenotype information of the patient (covered in the “understanding Huntington disease’ section) and the other is sequencing data that tell you if the patient carries 36 or more CAG repeats in a certain region of the HTT (covered in the previous section).
Back to the beginning; DNA was extracted from the blood sample of a patient with ataxia. Ataxia can be associated with Huntington disease since it indicates impaired coordination of movement. Also, exome capture was performed, and the captured exome was sequenced using an Illumina sequencing platform. Sequencing data were analyzed. Then, variants were annotated and prioritized by EVIDENCE 6, which were then curated by our clinical team. It was discovered that the patient had 37 pathogenic repeats with reduced penetrance. Therefore, she was diagnosed with Huntington disease.
Some limitations that are worth mentioning were faced in diagnosing repeat expansion disorders.
First, for WES, repeats on non-coding regions cannot be sequenced, thus, cannot be diagnosed. Fragile X syndrome is a good example. A repeat of the fragile X syndrome is located on the 5’ UTR of gene FMR1, which cannot be targeted by the capture kit for WES.
Second, the sequencing platform used produces reads that are 150 bp long, which is unable to precisely count repeats longer than 150 bp. Therefore, since the number of repeats provided was estimated, we recommend undergoing a confirmatory clinical test.
Currently, WES at 3billion, inc. can only diagnose 10 repeat expansion disorders, including Huntington disease. In Conclusion, due to the aforementioned limitations, undergoing confirmatory clinical tests, such as whole-genome or long-read sequencing, for repeat expansion disorders is recommended.
References
- Dayalu P, Albin RL. Huntington disease: pathogenesis and treatment. Neurol. Clin. 2015. PMID: 25432725
- Weber JL, Wong C. Mutation of human short tandem repeats. Hum. Mol. Genet. 1993. PMID: 8401493
- Depienne C, Mandel JL. 30 years of repeat expansion disorders: What have we learned and what are the remaining challenges?. Am. J. Hum. Genet. 2021. PMID: 33811808
- Caron NS, Wright GEB, Hayden MR. Huntington Disease. In: Adam MP, Mirzaa GM, Pagon RA, et al., eds. GeneReviews®. PMID: 20301482
- Walker FO. Huntington’s disease. Lancet. 2007. PMID: 17240289
- Seo GH, Kim T, Choi IH, et al. Diagnostic yield and clinical utility of whole exome sequencing using an automated variant prioritization system, EVIDENCE. Clin. Genet. 2020. PMID: 32901917
Get exclusive rare disease updates
from 3billion.

Yongjun Song
Medical geneticist skilled in variant interpretation. Mainly interested in developing interpretation software including pipeline for rare disease diagnostics





