Limitation of NGS: Spinal Muscular Atrophy (SMA) Genetic testing
Owing to the combined technology of massive parallel sequencing and bioinformatic analysis in a short time, the next-generation sequencing (NGS), has been widely used for disease diagnosis, prognosis, therapeutic decision, and follow-up of patients replacing the traditional genetic testing such as single gene testing using Sanger sequencing. Despite the fact that NGS has the strong points like ultra-high throughput with less DNA, scalability, and speed, it has still its own limitations. The limitations of exome sequencing1 and genome sequencing have already been described2, but here the limitation of NGS will be mainly focused by discussing how difficult it is to achieve the molecular diagnosis of spinal muscular atrophy with NGS.
What is Spinal Muscular Atrophy?
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disorder affecting the motor neurons that cause muscle weakness or atrophy. For most affected individuals, onset ranges from infancy to early childhood; however, late adult onset has also been reported. Although SMA is divided into several different clinical subtypes depending on the age of onset and maximum motor function attained, molecular genetics of SMA are the same and phenotypes are in a continuous spectrum.3 Early and accurate diagnosis of SMA is extremely critical compared to other rare genetic disorders, as a couple of treatment options are available, one of which is the newly approved ‘Zolgensma’ that treats SMA patients with improved prognosis as a one-shot gene therapy.4
What causes Spinal Muscular Atrophy?
SMA is caused by biallelic loss of the SMN1 gene in 5q13 encoding for SMN protein that is known to play a pivotal role in the survival of motor neurons. The loss of these neurons located in the spinal cord results in the interruption of signaling between the brain and muscles. The molecular diagnosis of SMA requires the identification of two pathogenic variants that cause loss of SMN1. While most patients have homozygous whole gene deletion of SMN1, a small fraction of patients has a compound heterozygous variants of CNV deletion and pathogenic single nucleotide variant or small insertion/deletion variant (SNV/INDELs).3
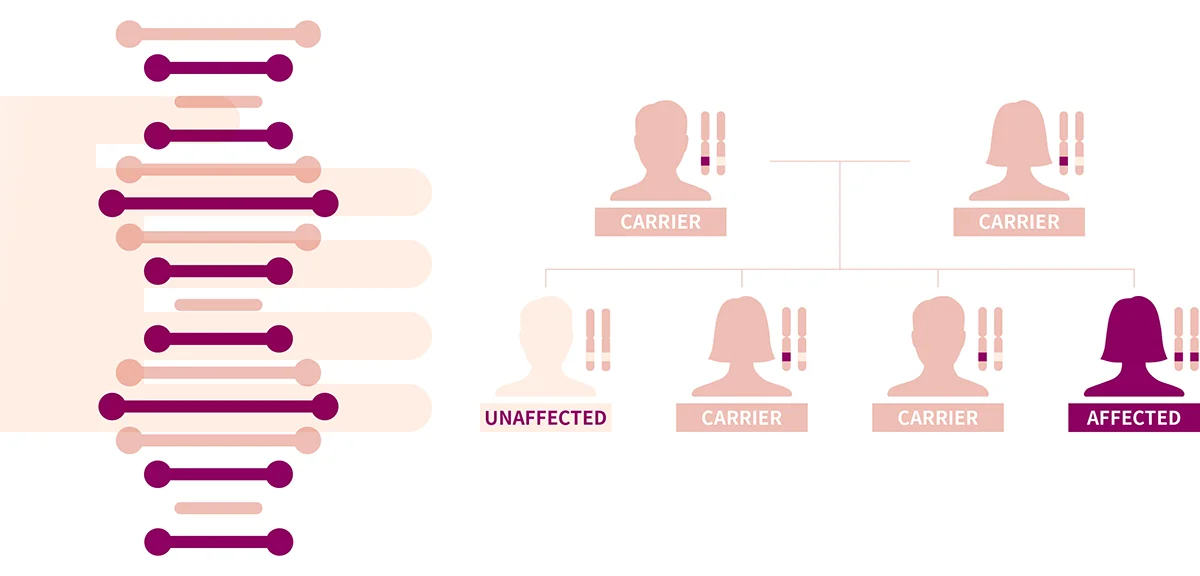
Interestingly, the presence of a disease-modifying gene SMN2 make the molecular diagnosis of SMA complicated. SMN2 is a ‘backup’ copy of SMN1, therefore having almost identical sequences to SMN1. Both genes, SMN1 and SMN2 are capable to produce SMN protein. However, SMN2 has a translationally silent variant c.840C>T in exon 7 that splicing. As a result of this variant, the c.840C>T variant, SMN2 predominantly generates a non-functional copy of SMN protein called SMNΔ7, which lacks exon 7. Approximately 80% of products derived from SMN2 are non-functional SMNΔ7, and remaining 20% are normal SMN proteins. It implies that the quantity of SMN2 is associated with the severity of the disease.5
The Limitation of next-generation sequencing
The presence of SMN2 imposes a significant challenge in sequencing these genes by the NGS technology. Because sequences for SMN1 and SMN2 are nearly identical, sequence reads from SMN2 are indistinguishable from SMN1. Therefore, NGS sequencing of SMN1 and SMN2 is difficult as sequencing reads for either SMN1 or SMN2 can be aligned to each other. Current NGS technology primarily produces a sequencing length of about ~150 bp which is not enough to distinguish sequencing reads from SMN1 and SMN2. As this is a technical limitation of current NGS technology, all diagnostic sequencing that utilizes short-read sequencers also has similar limitations and can only be avoided by performing long-read sequencing or non-NGS-based technology. Therefore, genetic testing for patients who are suspected of SMA should first be tested using non-NGS technology such as long-range PCR that specifically targets SMN1 or long-read sequencing. In practice, multiplex ligation-dependent probe amplification (MLPA), which is sequencing independent, is the most common technique that can reliably detect SMN1 deletion, while long-range PCR coupled with Sanger sequencing can reliably detect any heterozygous SNV/INDELs that might be present.3
Although it has limitations, it is not impossible to utilize NGS to diagnose SMA . Because of c.840C>T in exon 7 of SMN2, it is possible to distinguish sequence reads that came from SMN1 and SMN2 at exon 7. Therefore, SMN1 deletion can be detected by monitoring a loss of SMN1 exon 7 coverage. As homozygous deletion of SMN1 is the most frequent cause of SMA, NGS can be used to diagnose SMA patients. Nonetheless, for patients suspected of SMA, it is best practice to first perform MLPA-based tests for SMN1 deletion, then perform targeted gene tests for SMN1 if MLPA results in negative or inconclusive.

Curious How We Boost Diagnostic Accuracy?
Get the newsletter every month and discover the breakthroughs with the latest reanalysis report.
Conclusion
With the growing accessibility of NGS, exome and genome sequencing are now becoming first-tier genetic testing tools for all patients potentially suffering from genetic disorders. 3billion not only does our best to provide and update accurate and prompt information on such limitations of NGS to physicians and patients but also makes an effort to overcome those technical limitations by investigating.
References
- https://3billion.io/blog/limitations-of-whole-exome-sequencing/
- https://3billion.io/blog/limitation-of-whole-genome-sequencing
- Hassan, H.A., Zaki, M.S., Issa, M.Y. et al. Genetic pattern of SMN1, SMN2, and NAIP genes in prognosis of SMA patients. Egypt J Med Hum Genet 21, 4 (2020).
- https://www.zolgensma.com/
- Prior TW, Leach ME, Finanger E. Spinal Muscular Atrophy. 2000 Feb 24 [Updated 2020 Dec 3]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023
Get exclusive rare disease updates
from 3billion.

Seung Woo Ryu
Medical geneticist focused on clinical molecular genetics for rare diseases, dedicated to delivering accurate and accessible diagnostics to patients in need.





