How can you diagnose mitochondrial disease using WES and WGS?
What is mitochondrial disease?
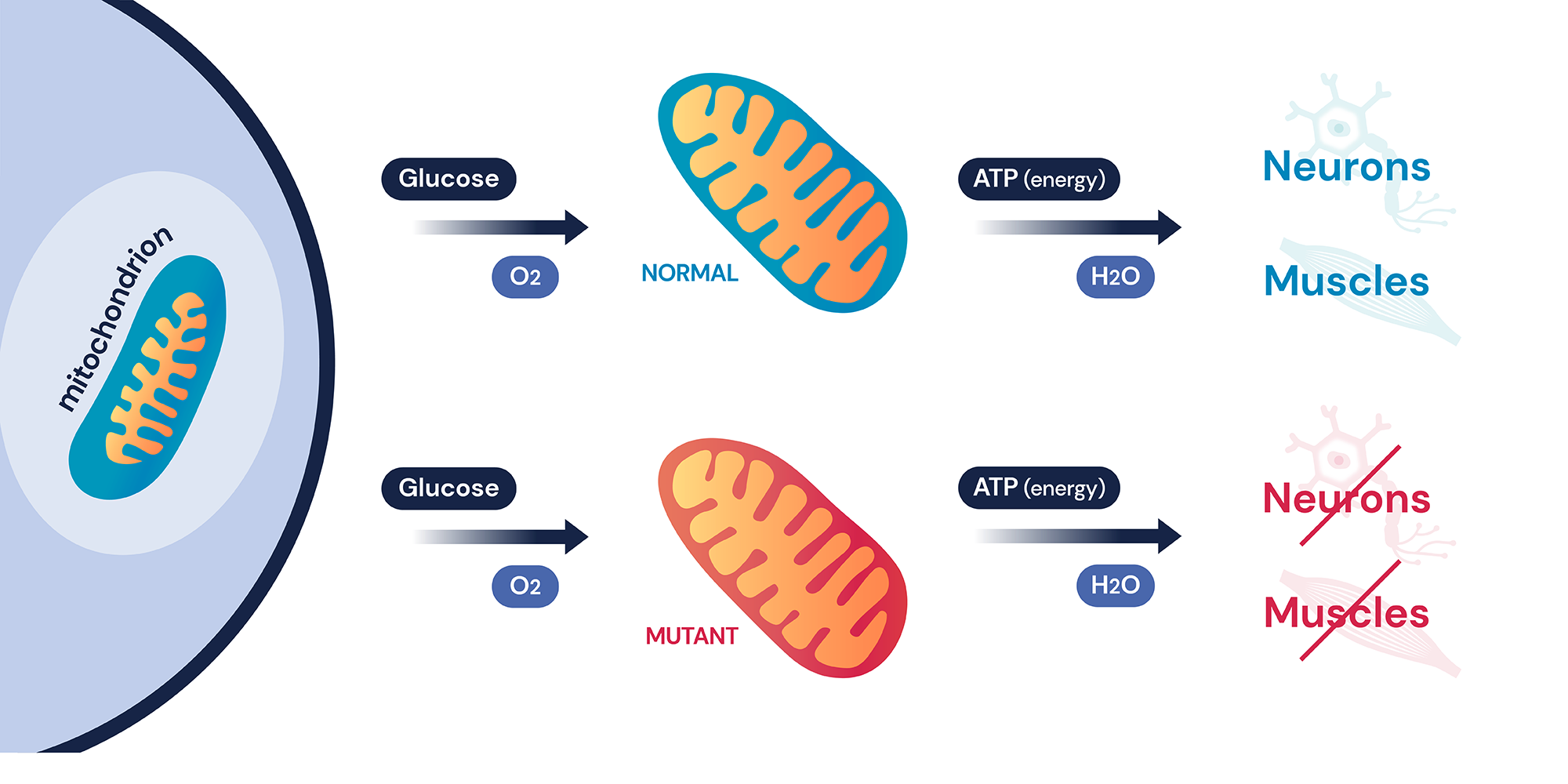
Mitochondria is a small worm-like organelle that performs cellular respiration called oxidative phosphorylation. Oxidative phosphorylation is a complex biochemical pathway that produces the majority of cellular ATP used by a cell using a series of protein complexes and small molecules called electron transport chains (ETCs)1. Therefore, dysfunction in mitochondria often affects the two most energy-demanding areas of the body: neurons and muscles, causing a series of diseases known as mitochondrial disorders. Mitochondrial disorders are known for their clinical and genetic heterogeneity and are often difficult to diagnose due to their wide clinical manifestation and symptoms. They can be as simple as non-syndromic optical neuropathy or hearing loss to more severe syndromic neuromuscular symptoms that can have a detrimental effect on patients’ health1, 2. As most mitochondrial disorders are progressive and the most severe disorders often have early onset, it is especially important to get the correct diagnosis at an earlier age.
Diagnosis of mitochondrial disorders is difficult and requires a detailed examination of the patient’s symptoms by a physician in the field. Furthermore, specialized biochemical, molecular, and tissue examinations are required for a correct diagnosis. The patients often go through a long extensive diagnostic journey including multiple clinicians and genetic panel testing before they receive a correct diagnosis. However, with increased accessibility of the whole exome and genome sequencing, patients can now receive a genetic test that searches for the entire human genome. Mitochondrial disorders, mostly associated with a genetic cause, can also benefit from WES/WGS as a first-tier test. Let’s find out how molecular diagnosis of mitochondrial disorders is made in clinical practice.
Mitochondrial DNA variants in clinical genetics
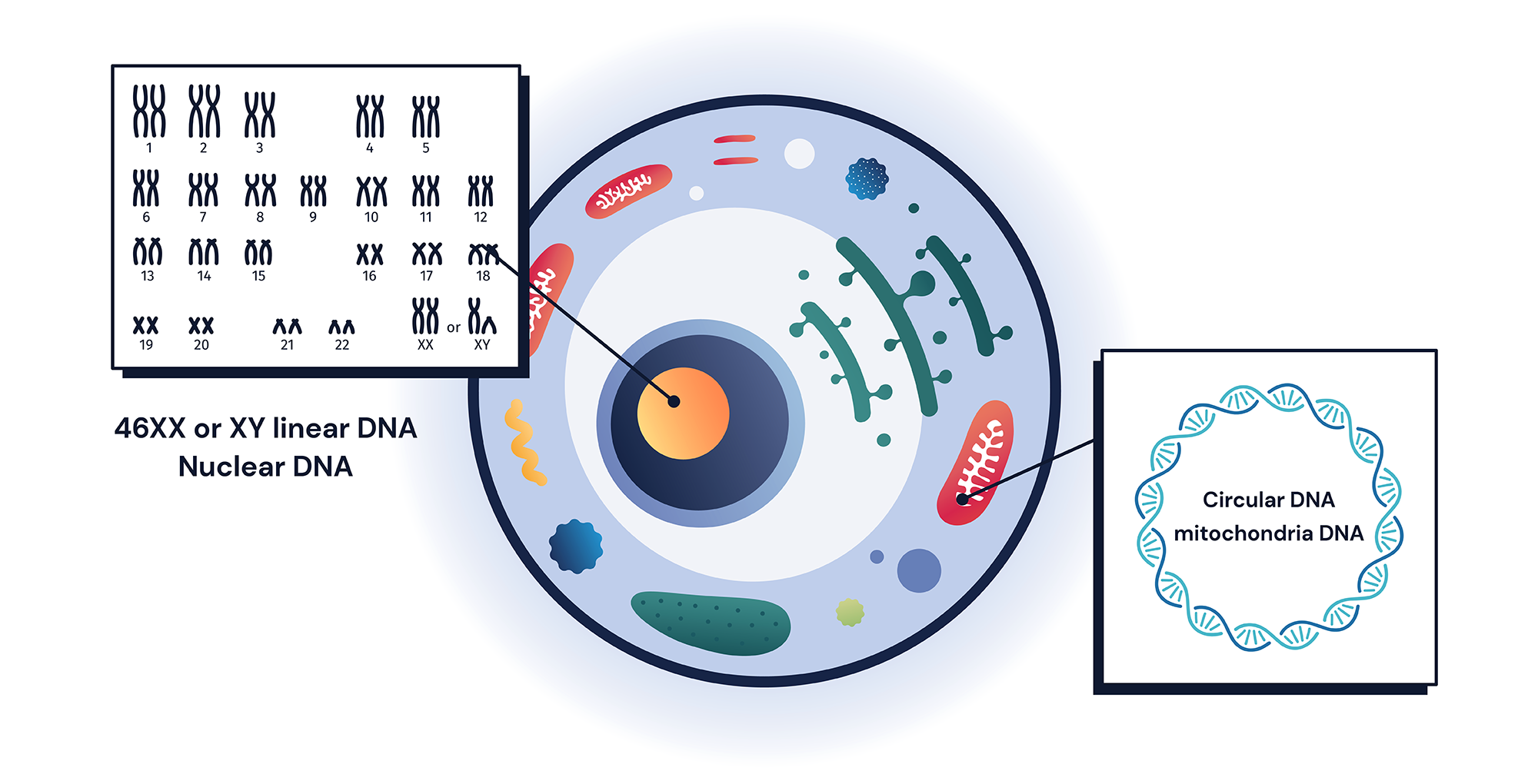
The human genome consists of 23 different chromosome pairs where each copy is inherited from each parent. However, the human genome also includes extra copies of a smaller-sized genome called mitochondrial genome (mtDNA)1. mtDNA is much smaller in size: only 16kb. It contains 37 different genes that lack intron, and are transcribed and translated within mitochondria3. Genes code for 13 different ETC proteins and include 2 ribosomal RNAs and a full set of 22 tRNAs, used for their own protein translation3.
Changes to mtDNA can sometimes cause serious damage to mitochondrial function and several rare human diseases are known to be caused by mtDNA variants1, 4. mtDNA is one of the most gene-dense DNA in humans, therefore, variants are often expected to be more pathogenic than variants from nuclear DNA2, 3, 5. Additionally, any potentially pathogenic changes to mtDNA non-coding RNAs: tRNA, and rRNA, often cause negative impacts on mitochondrial function5.
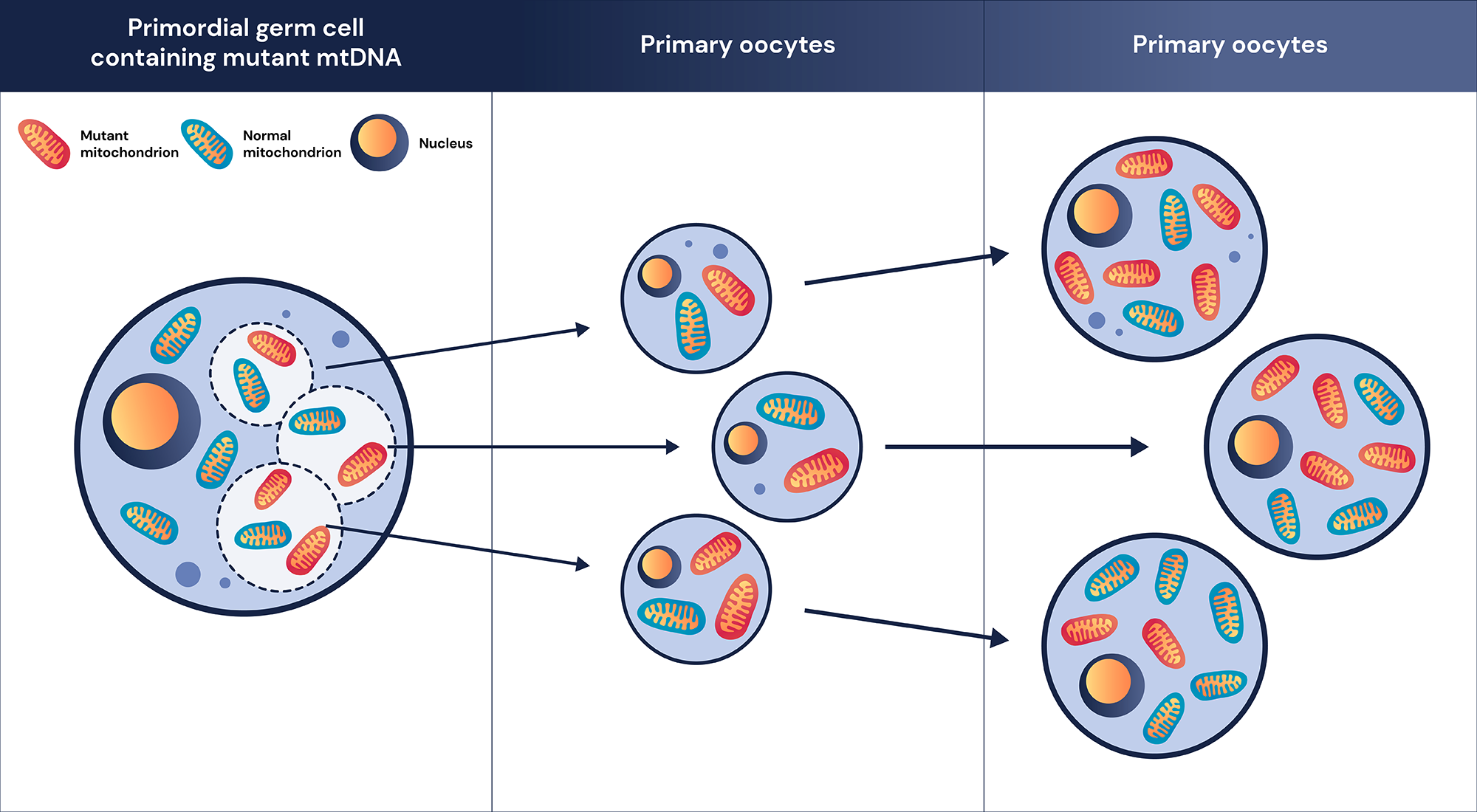
Variants from mtDNA are unique in that there are several unique features to consider from a clinical genetics point of view that is different from other nuclear variants. First, mtDNA copies are present in more than two copies. All other human DNAs are present in pairs, where each copy is inherited from single parents5. However, there is more than one copy of mtDNA per cell. In fact, there can be a few hundred to thousands of mtDNA per cell depending on cell types and sizes, a phenomenon called mtDNA polyploidy5, 6. Therefore, when we describe variants from mtDNA, we must use different zygosity terms other than heterozygous (one copy has variant)/homozygous (both copies have variant). For mitochondrial variant, we use percent heteroplasmy (% copies that have variant) and homoplasmy (100% copies have variant). A higher heteroplasmic percent of a potentially pathogenic variant often increases the disease’s burden for patients6–8.
Second, during mitosis, all nuclear DNAs are divided equally between two daughter cells. Therefore, each of the daughter cells maintains its variant zygosity after division1. However, mtDNA does not follow the same replication during cell division as mtDNAs are distributed randomly during cell division 6, 9. This creates situations where heteroplasmic alleles can shift in percentage during cell division. This phenomenon is called replicative segregation and can also happen during meiosis 1, 9. Therefore, even within the same individual, depending on the tissue and cell type, a different percentage of a heteroplasmic allele can present. Patients’ symptoms can also change as a result of replicative segregation as the heteroplasmic allele percentage of the pathogenic variant could be different throughout the body 6-9. Together with polyploidy, mitochondrial replicative segregation brings unique challenges to the interpretation of mitochondrial variants.
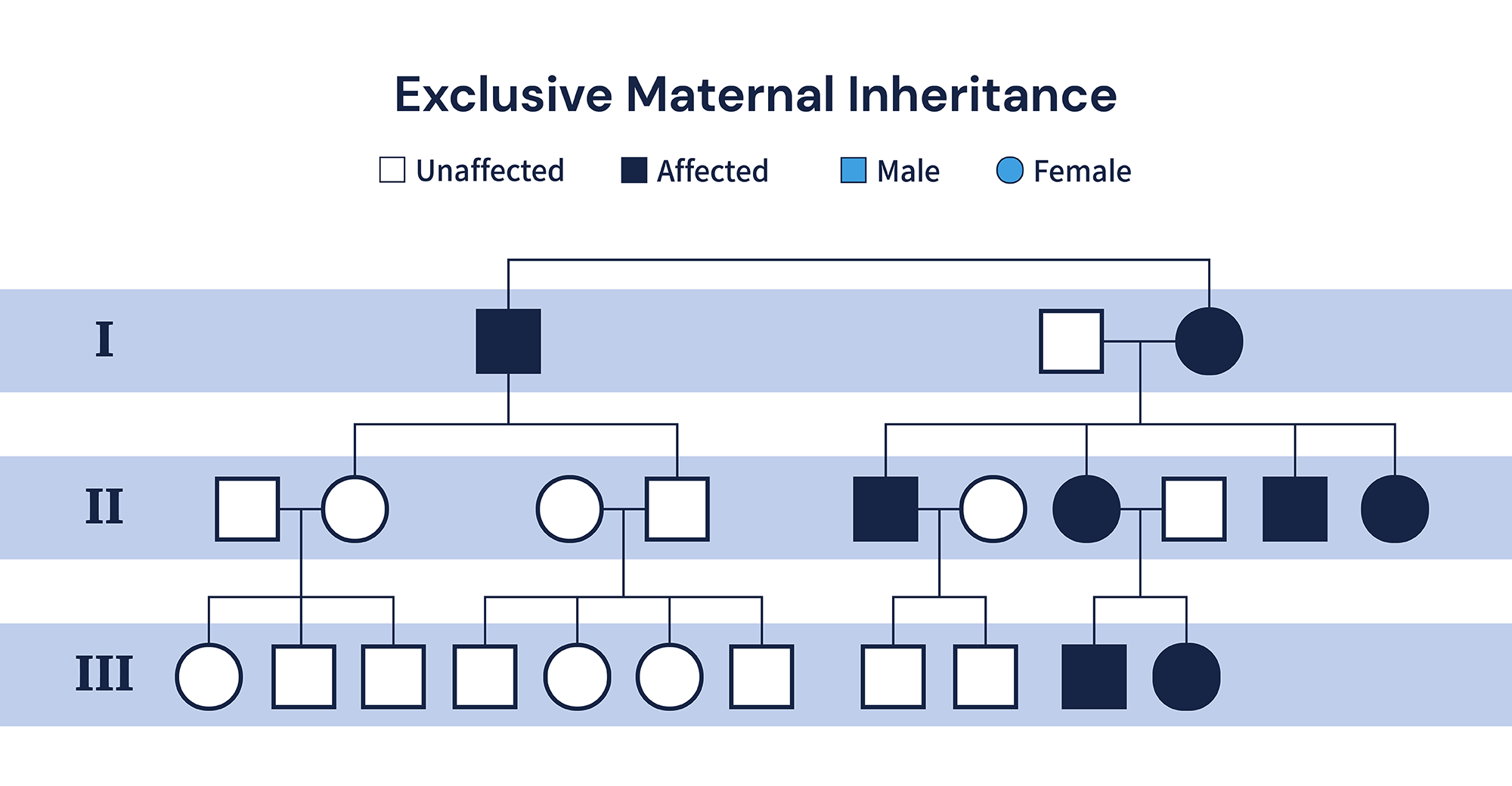
Third and finally, mtDNA is inherited exclusively from the mother. During the fertilization process, only mitochondria from the ova are left present in the zygote1. Therefore, a family test is especially important for the diagnosis of pathogenic mitochondrial variants as the proband’s mother or siblings might carry the same mitochondrial variant with a milder phenotype or in lower heteroplasmic percentage due to replicative segregation 6-9.
Real patient case M
Let’s take a look at how these characteristics of the mitochondrial genome affect a variant interpretation during the molecular diagnosis process in a clinical laboratory setting. M is an adolescent southeast Asian male patient who suffers from neuromuscular symptoms including muscle weakness, seizure, hearing loss, and encephalopathy. Whole-exome sequencing test was ordered to identify the potential molecular cause of his symptoms and a sample of his blood was given to us for the testing. (For more information about samples we accept and their process, please visit our product and blog pages 10-12) However, we did not find any potential pathogenic variant that could explain his symptoms from the nuclear genome.
The mitochondrial genome is not directly targeted by whole-exome sequencing but can be sequenced using off-target reads as they are smaller in size and abundant in a sample. From the mtDNA read, our clinical team has successfully identified an MT-3243A>G substitution variant. MT-3243A>G variant is located at the mitochondrial gene, MT-TL1, which codes for tRNA for leucine (UUR) and is known to be one of the most common causes of a mitochondrial disease called MELAS 6-13. MELAS stands for Mitochondrial Encephalopathy with Lactic Acidosis and Stroke-like episodes. MELAS is a genetically heterogeneous disorder (which means there is more than one gene that can cause MELAS) and has a highly variable phenotype with multiple systemic symptoms ranging from neurological to metabolic dysfunctions 13-15.
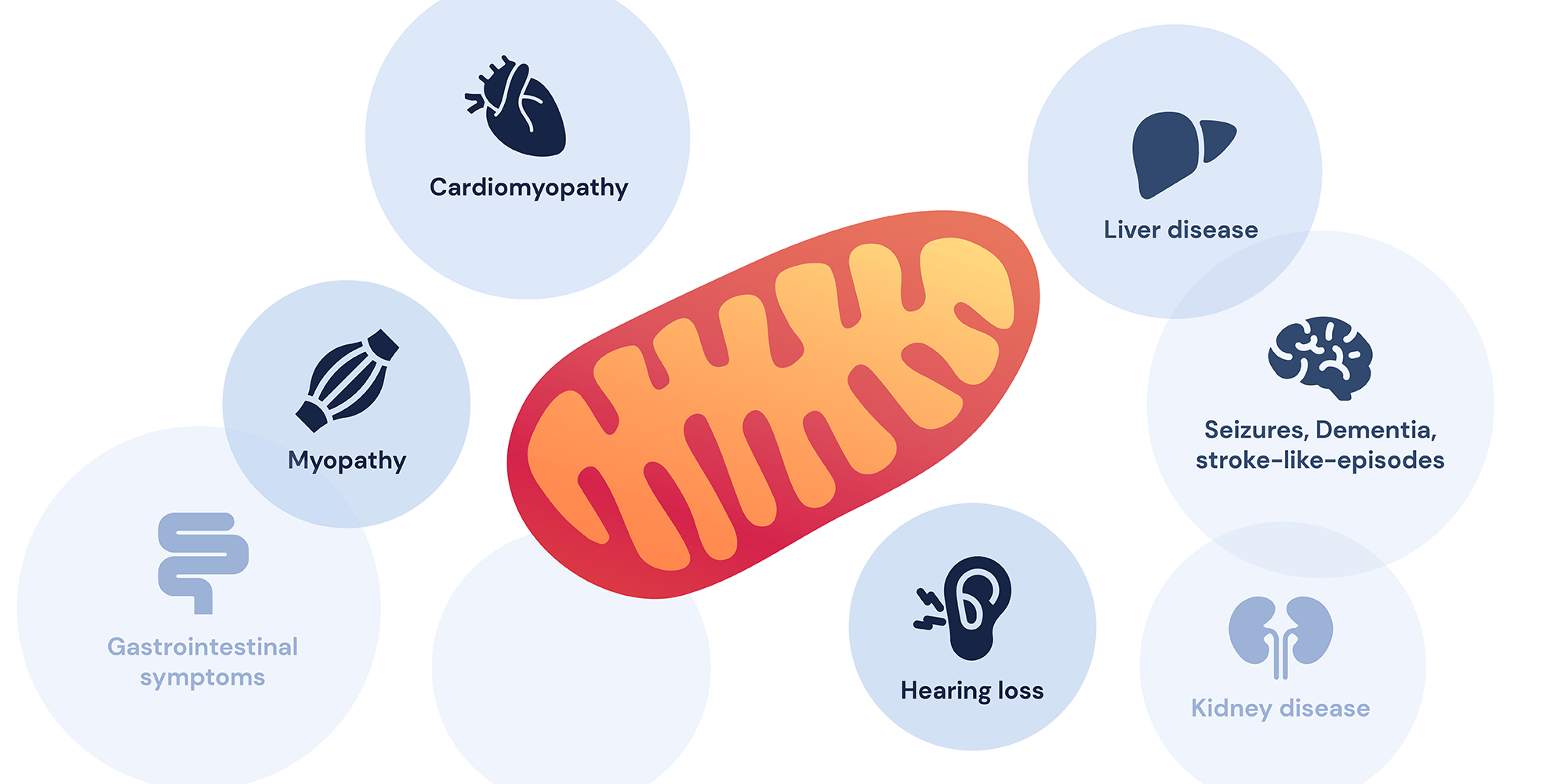
The most frequent symptoms of MELAS are hearing loss, generalized seizures, muscle weakness, and stroke-like episodes. Additional symptoms could include optical neuropathy and ataxia which could explain our patient’s symptoms13. In order to classify the MT-3243A>G variant via ACMG guidelines (For more information on ACMG rules, please visit our blog post on ACMG guidelines16), our clinical team first evaluated the allele frequency of the variant from a general population 6-8. From gnomAD, an open-source database that collected over 200,000 healthy individuals’ exome or genome data17, 6 different healthy individuals were identified where 5 individuals were found to have around 10-20% heteroplasmy while one individual had about 40-50% heteroplasmy. No homoplasmic individuals were identified indicating that this variant is extremely rare and further emphasizing that MT-3243A>G could be pathogenic (ACMG rule PM2). In silico tools, (a computational prediction of pathogenicity) also predicted that MT-3243A>G also has a MitoTIP score of 13.3 (likely pathogenic) and HmtVar score of 1.00 (pathogenic) (ACMG rule PP3) 18. Clinvar database, genomic variant database, and its relationship to human health, also concluded that this variant is pathogenic and was identified in multiple individuals that suffer from MELAS (ACMG rule PS4 and PP5) 19. In addition, our clinical team identified that MT-3243A>G variant has been seen to segregative with diseases in multiple affected individuals from the same family (ACMG rule PP1) 15, while a change in the same nucleotide residue changes to different nucleotide MT-3243A>T is also known to be pathogenic variant (ACMG rule PM5). Together, this information suggests that the MT-3243A>G variant is pathogenic, and can explain the symptoms. However, the patient’s sequencing results showed that only 32% of his mtDNA carried this variant, indicating that the variant is present in a heteroplasmic state. Therefore, our clinical team had to assess if 32% heteroplasmic variants can explain the patient’s phenotype.
According to a paper summarized by Dr. Dvorakova et al in 2016 “The phenotypic spectrum of fifty Czech m.3243A>G carriers” who looked at heteroplasmy level of MT-3243A>G in 50 MELAS patients, he and his colleague have discovered that heteroplasmy of MT-3243A>G from patients blood could be as low as 8%, but often higher in affected tissue 14. Since our clinical team could not analyze the same from the patient’s affected tissue sample, the team decided to release inconclusive results as validation is needed with a different technique that targets mitochondria in affected tissue samples. (for more information about our report please visit our product page 20)
Mitochondrial disorders in WES/WGS setting
Mitochondrial genomic variants, despite being small, account for about 0.4% of our total diagnosis yield so far. Because mitochondrial disorders are highly heterogeneous while symptoms associated can be wide and variable, patients often have a very difficult time getting the right diagnosis for their symptoms. The diagnostic odyssey of patients with mitochondrial disorders on average consists of about 8 different clinicians, while about 55% of the time their first diagnoses were incorrect 21. This is a bit higher than the average 7 clinicians and 44% misdiagnosis rates compared to other rare diseases, indicating that mitochondrial disorders are harder to diagnose compared to other rare genetic disorders 22.

Access Our Webinars on WGS and WES
Watch our expert-led sessions on how WGS and WES are revolutionizing the diagnosis of rare diseases.
Even in today’s example MT-3243A>G variant, which is one of the most frequent causes of MELAS syndrome, often has variable expressivity from complex neuromuscular symptoms like patient M, to milder symptoms such as non-syndromic hypertrophic cardiomyopathy or proteinuria 15, 23, 24. Therefore, many doctors and primary care physicians often have trouble referring patients to the correct specialist. Making matters more complex, over 250 genes from nuclear genomes also are thought to be related to mitochondrial disease and this number is growing continuously making it harder for physicians to keep up with the latest research for a small number of patients. Further complicated by patients who are mistakenly thought to have mitochondrial disorders.
Only 11% of suspected mitochondrial patients receive a diagnosis by conventional sanger-based approach 25, 26, while targeted gene panels are usually about 22%~24% demonstrating limitations of traditional panel-based approach while clinicians/patients have to perform multiple panel sequencing increasing burden for patients and society 25, 27, 28. From the next generation whole exome sequencing-based approach, numbers can be as high as 68% strongly suggesting that WES should be the first to-go genomic tests for anyone who is suspected of having a mitochondrial disorder 25. Because mitochondrial disease can be caused by both nuclear and mitochondrial genome, patients could benefit from WES test which sequences both nuclear and mitochondrial genome. However, several limitations do exist. Because most WES, including WES from 3billion, does not target the mitochondrial genome as a primary target, most sequencing reads are made by off-target reads. Most of the time, off-target reads do have sufficient read depth, but this also means sometimes coverage is not high enough for WES to detect mtDNA variants, especially low heteroplasmy variants. To increase the detection yield, some labs use exome capture kits in combination with mitochondrial prob in order to increase mitochondrial coverage depth. WGS also solve this limitation as mtDNA coverage is often as high as 1000x. Patients with mitochondrial disorders need to be validated by tissue-specific biochemical testing in order to correctly diagnose a patient. Similar to our patient M, because samples we receive are not from the affected area of tissue, it is difficult to assess the actual level of heteroplasmy at the affected tissue. Nonetheless, even with such limitations, unbiased WES tests could be a powerful tool for patients who are suffering from obvious mitochondrial disorders 25, 28-30.
References
- Nussbaum RL, McInnes RR, Willard HF. Thompson & Thompson Genetics in Medicine. Eighth edition. Elsevier; 2016.
- Lawless C, Greaves L, Reeve AK, Turnbull DM, Vincent AE. The rise and rise of mitochondrial DNA mutations. Open Biol. 2020;10(5):200061.
- Bernardino Gomes TM, Ng YS, Pickett SJ, Turnbull DM, Vincent AE. Mitochondrial DNA disorders: from pathogenic variants to preventing transmission. Hum Mol Genet. 2021;30(R2):R245-R253.
- Moggio M, Colombo I, Peverelli L, et al. Mitochondrial disease heterogeneity: a prognostic challenge. Acta Myol. 2014;33(2):86.
- Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6(5):389-402.
- Wong LJC, Chen T, Wang J, et al. Interpretation of mitochondrial tRNA variants. Genet Med. 2020;22(5):917-926.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-423.
- McCormick EM, Lott MT, Dulik MC, et al. Specifications of the ACMG/AMP standards and guidelines for mitochondrial DNA variant interpretation. Hum Mutat. 2020;41(12):2028-2057.
- Zhang H, Burr SP, Chinnery PF. The mitochondrial DNA genetic bottleneck: inheritance and beyond. Essays Biochem. 2018;62(3):225-234.
- Juan Alberto Aguayo. How do you decide between blood or buccal swab collection? 3billion Blog. Published December 7, 2021. Accessed April 12, 2022.
- OMIM Entry – #540000 – MITOCHONDRIAL MYOPATHY, ENCEPHALOPATHY, LACTIC ACIDOSIS, AND STROKE-LIKE EPISODES; MELAS. Accessed April 12, 2022.
- Dvorakova V, Kolarova H, Magner M, et al. The phenotypic spectrum of fifty Czech m. 3243A> G carriers. Mol Genet Metab. 2016;118(4):288-295.
- Finsterer J, Laccone F. Phenotypic heterogeneity in 5 family members with the mitochondrial variant m. 3243A> G. Am J Case Rep. 2020;21:e927938-1.
- Yeonho Kang. What are the ACMG Standards and Guidelines and how do they work? Accessed April 12, 2022.
- Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-443.
- Lott MT, Leipzig JN, Derbeneva O, et al. mtDNA variation and analysis using mitomap and mitomaster. Curr Protoc Bioinforma. 2013;44(1):1-23.
- Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062-D1067.
- Product details of rare disease genetic test | 3billion. Accessed April 12, 2022.
- Grier J, Hirano M, Karaa A, Shepard E, Thompson JL. Diagnostic odyssey of patients with mitochondrial disease: Results of a survey. Neurol Genet. 2018;4(2).
- The rare disease diagnostic odyssey – Raconteur. Accessed April 12, 2022.
- Shand JA, Potter HC, Pilmore HL, Cundy T, Murphy R. Increased Peripheral Blood Heteroplasmy of the mt. 3243A> G Mutation Is Associated with Earlier End-Stage Kidney Disease: A Case Report and Review of the Literature. Nephron. 2020;144(7):358-362.
- Motlagh Scholle L, Zierz S, Mawrin C, Wickenhauser C, Lehmann Urban D. Heteroplasmy and copy number in the common m. 3243a> G mutation—A post-mortem genotype–phenotype analysis. Genes. 2020;11(2):212.
- Theunissen TE, Nguyen M, Kamps R, et al. Whole exome sequencing is the preferred strategy to identify the genetic defect in patients with a probable or possible mitochondrial cause. Front Genet. Published online 2018:400.
- Neveling K, Feenstra I, Gilissen C, et al. A Post-Hoc Comparison of the Utility of Sanger Sequencing and Exome Sequencing for the Diagnosis of Heterogeneous Diseases. Hum Mutat. 2013;34(12):1721-1726.
- Lieber DS, Calvo SE, Shanahan K, et al. Targeted exome sequencing of suspected mitochondrial disorders. Neurology. 2013;80(19):1762-1770.
- Calvo SE, Compton AG, Hershman SG, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012;4(118):118ra10-118ra10.
Get exclusive rare disease updates
from 3billion.

Seung Woo Ryu
Medical geneticist focused on clinical molecular genetics for rare diseases, dedicated to delivering accurate and accessible diagnostics to patients in need.





