Validation of poor-quality pathogenic variants from next-generation sequencing results
Although whole exome sequencing (WES) can efficiently cover more than 90% of the protein coding sequence (CDS), some cases make it very difficult to accurately detect variants in the CDS region via WES using short reads1,2. Thus, we would like to discuss one of those cases.
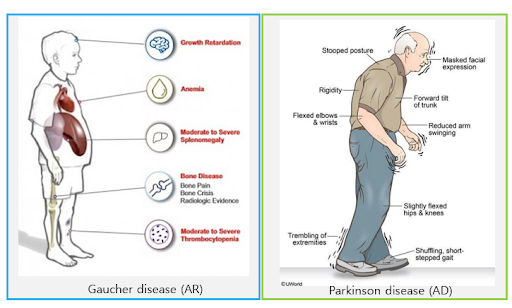
This patient, born in 1952, has shown recurrent falls, dyspnea, difficulty in raising his arms above his shoulders, and has recently been in a wheelchair. Based on the symptoms and the onset age, which was as an adult, neurodegenerative disorders were suspected. A well-fitted variant with high pathogenicity was identified from WES data. It was a heterozygous missense variant in the GBA gene3. This variant has been reported as pathogenic several times with sufficient evidence. Furthermore, several American College of Medical Genetics and Genomics pathogenicity rules were applied, which enabled the classification of the variant as pathogenic with a very high pathogenicity score4.
The GBA gene plays a crucial role in lysosomes making important enzymes and is known as a disease-relevant gene with autosomal recessive (AR) inheritance3,5. When both copies of the GBA gene contain pathogenic variants, it can lead to Gaucher disease, which is a severe, rare, inherited metabolic disorder3. Interestingly, some pathogenic variants in the GBA gene are known to increase the risk of suffering Parkinson’s disease with autosomal dominant (AD) inheritance3. We call these variants risk alleles, meaning not everyone with these alleles suffer from disorders5,6. Since common symptoms of Parkinson’s disease include tremors, slowed movement, rigid muscles, impaired posture, balance, etc., there seems to be no reason not to confirm this variant as a causal variant for this patient’s symptoms3,7. However, it was not as simple as it seemed.
As previously mentioned, the variant is known to be a risk allele for Parkinson’s disease when it is heterozygous. Therefore, the ratio of the reference allele to the alternate allele for heterozygous variants should be around 1:1. However, the observed ratio of this variant was around 3:1.
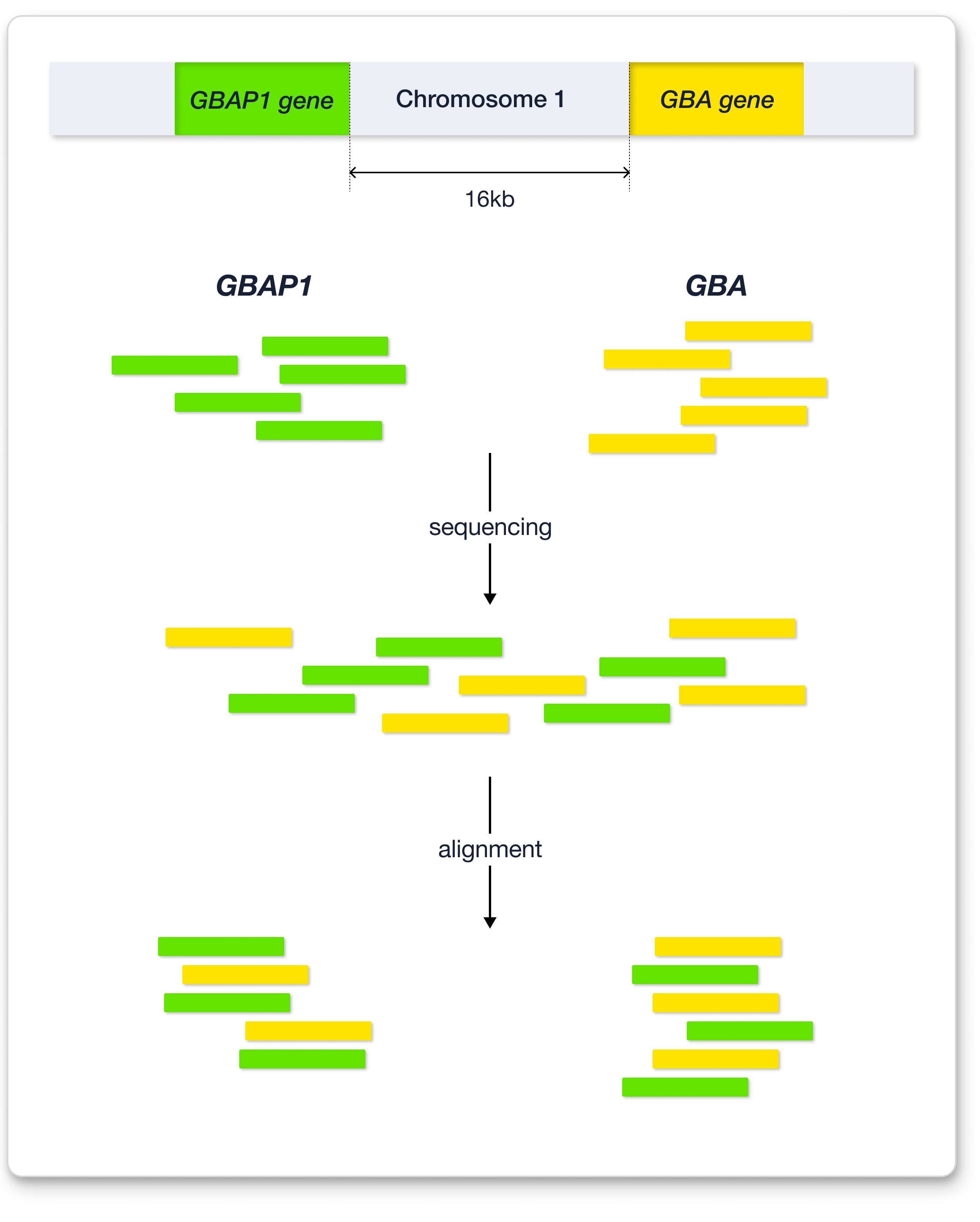
Several possibilities could have elicited this skewed ratio. Among them, it was the existence of highly homologous regions in the genome, also known as pseudogenes8. The meaning of “pseudo-“ is “fake.” Pseudogenes are genes that result from gene duplication but may have lost their functionality for some reason9. In other words, pseudogenes are rarely used for protein coding. The GBA gene has a known pseudogene called GBAP1, which is located close to GBA10. In most cases, the sequence of pseudogenes is similar to that of the original gene. In the case of GBAP1, the sequence homology with GBA is 96%–98%10. Since the WES platform we use generates reads with a size of only 150 bp, it becomes difficult to accurately align when the read originates from a gene that has a highly homologous pseudogene8. In other words, the presence of pseudogenes complicates the alignment process because the aligner cannot accurately map reads to either the original gene or the pseudogene.
In detail, during sequencing, DNA fragments from both GBA and GBAP1 are mixed, after which alignment occurs. Since the reads generated by our WES platform are short and the homology between GBA and GBAP1 is high, it becomes difficult to distinguish reads from GBA and GBAP1. Thus, some of the reads from GBA might be aligned to GBAP1. Likewise, some from GBAP1 might be aligned to GBA. Let’s say a variant is located on the GBA gene. While in alignment, some of the reads that span this variant position might map to GBAP1, implying that those reads were not properly mapped. To avoid this improper mapping, GBAP1 is masked, resulting in no mapping to GBAP1, and most reads from GBAP1 are mapped to GBA instead11. This process makes it impossible to distinguish whether the called variants in GBA are true or false. In other words, an alternative way to confirm the variant in the GBA gene needs to be considered. Therefore, Sanger sequencing was used12.
Sanger sequencing typically reads 500–600 bp of DNA fragments at a time. The alternative allelic depth was only 26% in the previous WES result. However, the alternative allelic depth from Sanger sequencing was approximately 50%, which strongly indicates that the variant is heterozygous.
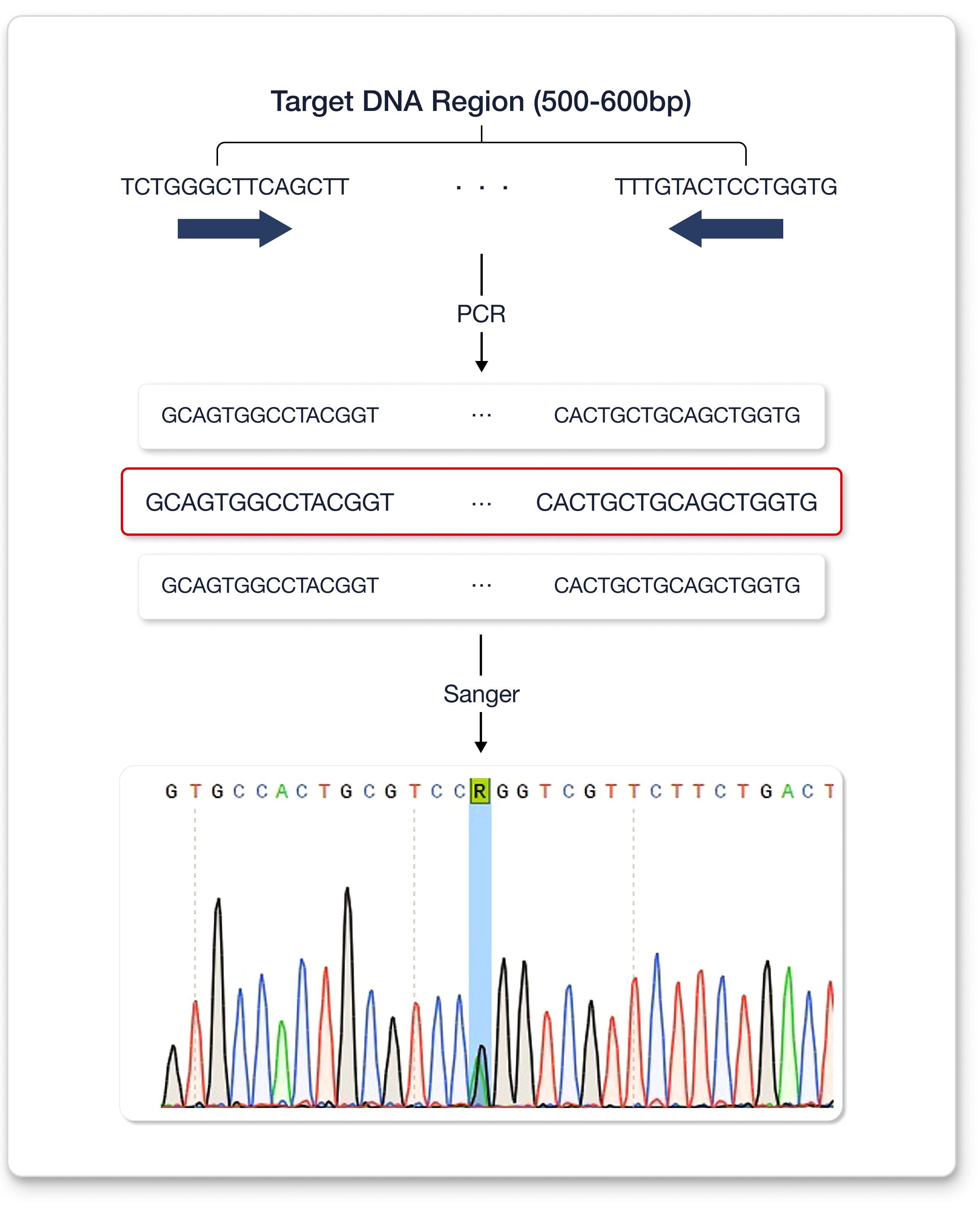
Summarily, detecting a pathogenic variant in GBA was difficult due to alignment issues caused by the presence of a highly homologous pseudogene. However, with the Sanger sequencing technique much longer DNA sequences can be read at once, enabling variant detection and shortening the patient’s diagnosis.
References
- Kendall JB, Joy DC, Lynette CR, et al. Limitations of exome sequencing in detecting rare and undiagnosed diseases. Am J Med Genet A. 2020; 182(6):1400-1406.
- Yury AB, Dmitrii EP, Andrey SG, et al. Systematic dissection of biases in whole-exome and whole-genome sequencing reveals major determinants of coding sequence coverage. Scientific Reports. 2020; 10:2057.
- Ellen S, Grisel L. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012; 11(11):986-998.
- McCormick EM, Lott MT, Dulik MC, et al. Specifications of the ACMG/AMP standards and guidelines for mitochondrial DNA variant interpretation. Hum Mutat. 2020; 41(12):2028-2057.
- Daphne EC, Jeroen VS, Joke AB, et al. Glucocerebrosidase: Functions in and Beyond the Lysosome. Journal of Clinical Medicine. 2020; 9, 736.
- KJ Billingsley, S Banderes, S Saez-Atienzar, et al. Genetic risk factors in Parkinson’s disease. Cell Tissue Res. 2018; 373(1):9-20.
- Micol A, Fabio B, Silvia C. Glucocerebrosidase defects as a major risk factor for Parkinson’s disease. Front. Aging Neurosci. 2020.
- Diana M, Ryan JS, Arunkanth A, et al. Navigating highly homologous genes in a molecular diagnostic setting: a resource for clinical next-generation sequencing. Genet Med. 2016; 1282-1289.
- Mighell AJ, Smith NR, Robinson PA, et al. Vertebrate pseudogenes. FEBS Lett. 2000; 468(2-3):109-14.
- Stefani Z, Silvia C, Bruno B, et al. GBA Analysis in Next-Generation Era: Pitfalls, Challenges, and Possible Solutions. The Journal of Molecular Diagnostics. 2017; 733-741.
- Ebbert M, Jensen TD, Jansen-West K, et al. Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight. Genome Biol. 2019.
- Sanger F, Nicklen S, Coulson R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977; 74(12): 5463-5467.
Get exclusive rare disease updates
from 3billion.

JiHye Kim
Medical geneticist with experience in various molecular works and specializing in variant interpretation for rare disease diagnostics





