Type I Neurofibromatosis / NF1 intron case
Patient H was a 6-year-old Korean boy who was clinically suspected of having type I Neurofibromatosis. He presented with general symptoms of type 1 neurofibromatosis which was the presence of Multiple café-au-lait spots and no other family members were affected. The patient was ordered a single gene sequencing test in a separate clinical laboratory in order to confirm the provisional diagnosis of type 1 neurofibromatosis. However, results came back negative with no pathogenic variants in the coding region of the NF1 gene. Therefore, physicians have requested whole genome sequencing testing from us for deep analysis. The test was ordered as a trio, but no other family member had a history of genetic illness.
As expected, no pathogenic or potentially pathogenic variants were observed from the patient’s coding region of the NF1 gene as well as other genes. Also, the patient did not carry pathogenic structural variants or copy number variants (SVs and CNVs). Upon further inspection, we have identified a potentially significant non-coding variant within the deep intronic region of NF1 between exon 14 and 15.

The variant NM_001042492.3(NF1):c.1642-449A>G (p.?) is located 449 nucleotides upstream of exon 15 and would not have been visible if the patient had performed WES or exomic gene panel testing instead of WGS.

WES: data of the same region from different patients. Note that no read is a converging variant of interest. Genomic position number varies slightly as WES was mapped to hg19 and WGS was mapped to hg38
The variant was identified to be de novo, as it was not identified in either the father or mother. The variant was not present in gnomAD 3.1.1, the largest freely available healthy population frequency data. Also, the variant has been previously reported in other NF1-related patients in the literature (PMID: 23668869).
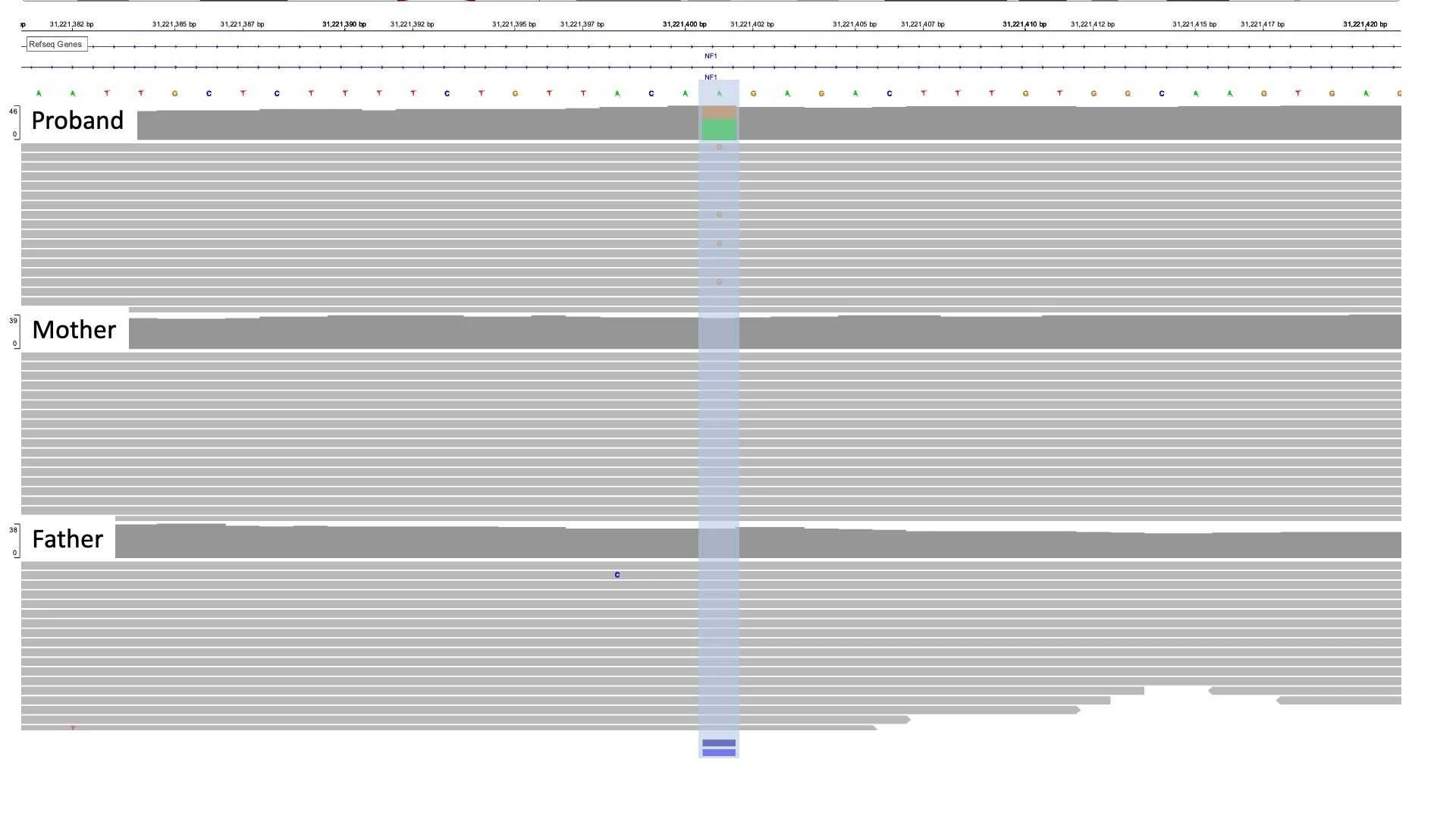
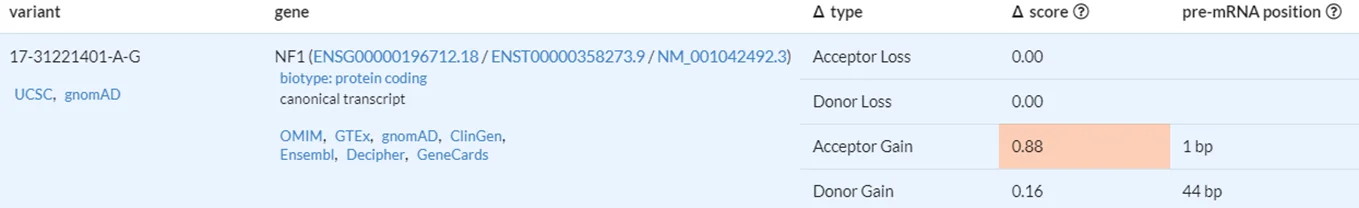
However because the variant is a deep intronic variant, it is difficult to predict the consequence of the variant at the protein level. Without experimental data such as RNA-sequencing or protein blots, we have to rely on computational prediction tools for analysis. SpliceAI, a deep learning-based splicing prediction tool from Illumina, has predicted that the NM_001042492.3(NF1):c.1642-449A>G (p.?) variant is highly likely to result in a change of splicing for the gene.
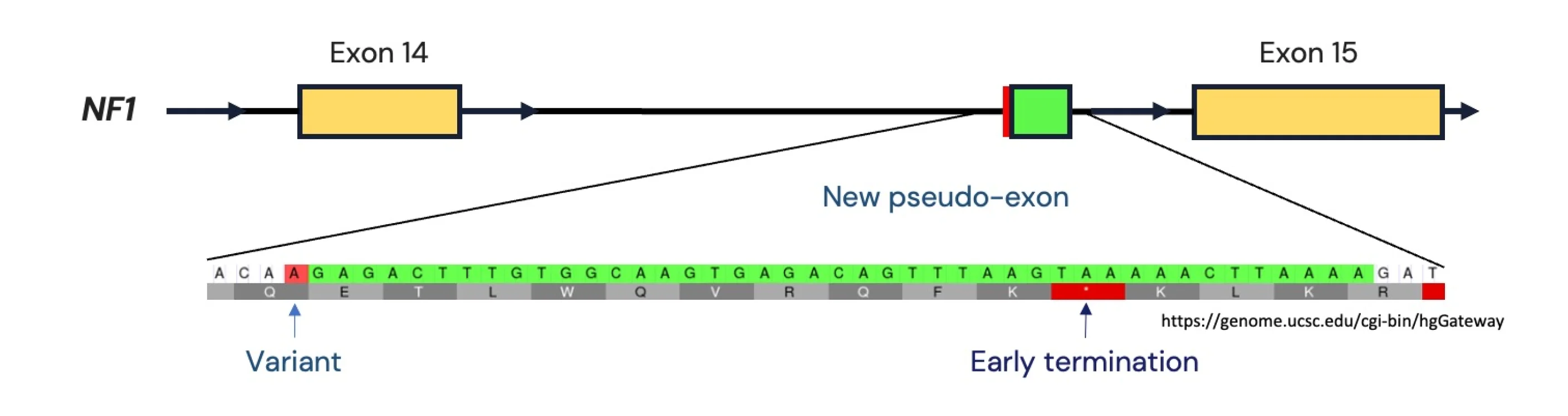
If the prediction is correct, this would result in the addition of 43 base pair pseudo-exon between exons 14 and 15. Because this pseudo-exon contains a premature termination codon, it would result in premature termination of the NF1 gene and result in a loss-of-function via nonsense-mediated decay.
Type I neurofibromatosis is an autosomal dominant single-gene disorder caused by a loss-of-function variant of the NF1 gene. Type I neurofibromatosis is one of the most common single-gene defects that affects about 1 in 3000 live births across the world. Patients with type I neurofibromatosis suffer from variable multisystemic abnormality consisting of multiple café-au-lait spots, axillary freckling, and most importantly, the formation of multiple tumors called neurofibromas. Neurofibromas are benign tumors from nerve tissues and can form anywhere in the body from the subcutaneous region to deep nodules. Most neurofibromas are benign but can develop into malignant tumors if left untreated. Early diagnosis is highly important as there are treatment options available. In 60~90% of suspected cases, a pathogenic variant can be identified in NF1 using single-gene tests including a whole-gene deletion. However, in the remaining cases, patients are left undiagnosed and have to rely on clinical findings for efficient diagnosis.
In conclusion, with the evidence provided, patient H was diagnosed with Type I neurofibromatosis from the pathogenic deep intronic variant in NF1. Since the variant is not part of the targeted region for WES, patients would have needed to get WGS in order to identify such variants. However, it is important to consider the limitations of WGS in identifying deep intronic variants. Because most additional variants (SNVs and small INDELs) that are identified from WGS compared to WES are part of non-coding regions such as intronic or intergenic regions, it is difficult to interpret the consequence as there is still a lack of knowledge about deep non-coding regions. Therefore, it is important for physicians to consider such limitations when ordering WGS tests.

What Makes Our Exome Testing Advanced?
Find out how our enhanced exome solutions deliver better coverage and accuracy.
If you are interested in the limitations of WGS:
For more information about type I neurofibromatosis:
Neurofibromatosis, type 1 (OMIM: 162200)
Get exclusive rare disease updates
from 3billion.

Seung Woo Ryu
Medical geneticist focused on clinical molecular genetics for rare diseases, dedicated to delivering accurate and accessible diagnostics to patients in need.




