










EVIDENCE
Automatic variant interpretation system, the core of 3billion variant interpretation technologyVariant Interpretation System, the answer to rare disease diagnosis
When a patient’s genetic information is analysed using whole genome sequencing (WGS) or whole exome sequencing (WES), approximately 100,000 up to millions of genetic variants are found, respectively.
These variants must be interpreted according to the variant interpretation guideline to identify pathogenic variants. It takes time to manually identify from 100,000 up to millions of variants, which may cause biased interpretation results. To solve this problem, 3billion has developed a variant interpretation system, EVIDENCE for accurate and fast diagnosis.
Why is EVIDENCE so unique?
We update our database with the newest information on a daily basisVariants detected by WGS or WES are filtered sequentially. To reflect the latest research, information from various databases is reviewed in this process.
Disease information
OMIM, Orphanet, HPOPathogenic variant information
ClinVar, HGMDPrediction
tools
Large-scale sequencing data
gnomAD, TopMed, in-house dataThe pathogenicity of over 100,000 variants can be automatically interpreted per minute
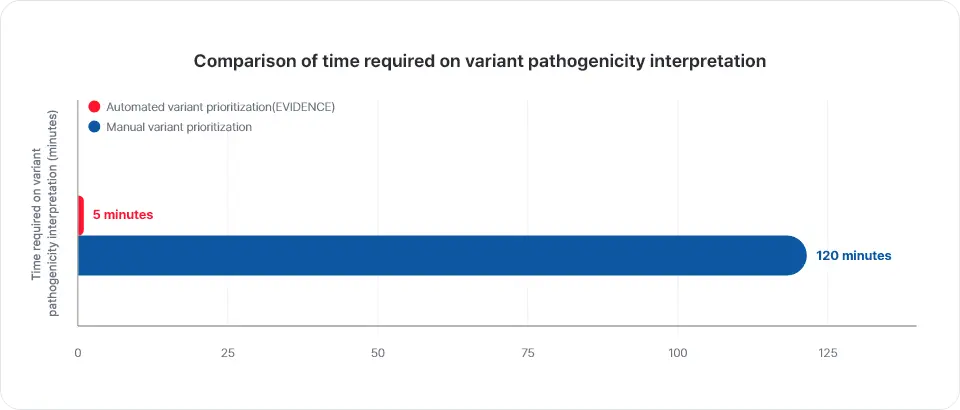
With EVIDENCE, variants can be interpreted in less than a minute, which is significantly faster than human interpretation. (*for WES) Compared with the variant analysis data of ClinVar, the sensitivity of EVIDENCE was 98.4% and the specificity was 99.9%, proving EVIDENCE’s superior variant interpretation performance. (3billion, 2021)


We convert the patient’s symptoms into quantifiable metrics, which are reviewed in the interpretation process
EVIDENCE curates a list of variants that are prioritized by relevance to the patient’s symptoms. EVIDENCE calculates a “symptom similarity” score to determine the similarity between known symptoms caused by the variant and the patient’s actual symptoms. This greatly increases the efficiency of variant prioritization.
We utilize our in-house database
3billion's Medical Genetics Division and Bioinformatics team are building a proprietary database of pathogenic variants, new diseases, and population frequencies that have not yet been found in public databases. This information plays an important role in increasing the sensitivity and specificity of EVIDENCE.
In addition, anonymized diagnostic results of patients are used to improve EVIDENCE.
How does EVIDENCE work?
EVIDENCE analyzes variants according to the ACMG Standards and Guidelines using both patient's genetic and symptom information. This process can be divided into the following six steps.